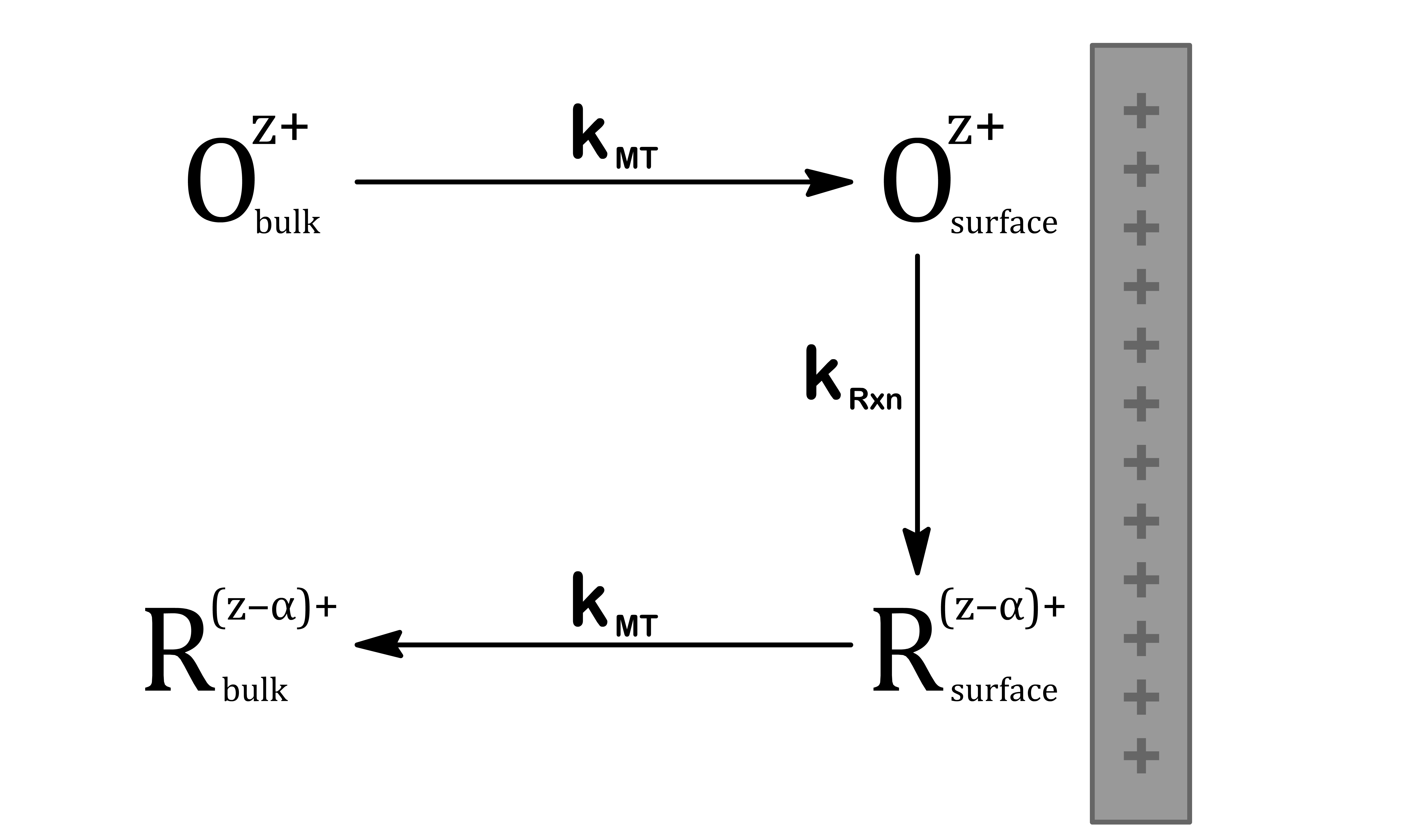Before we jump into the kinetics of electrochemistry, we shall try to develop a general and advance model for rates using the notion of transition states
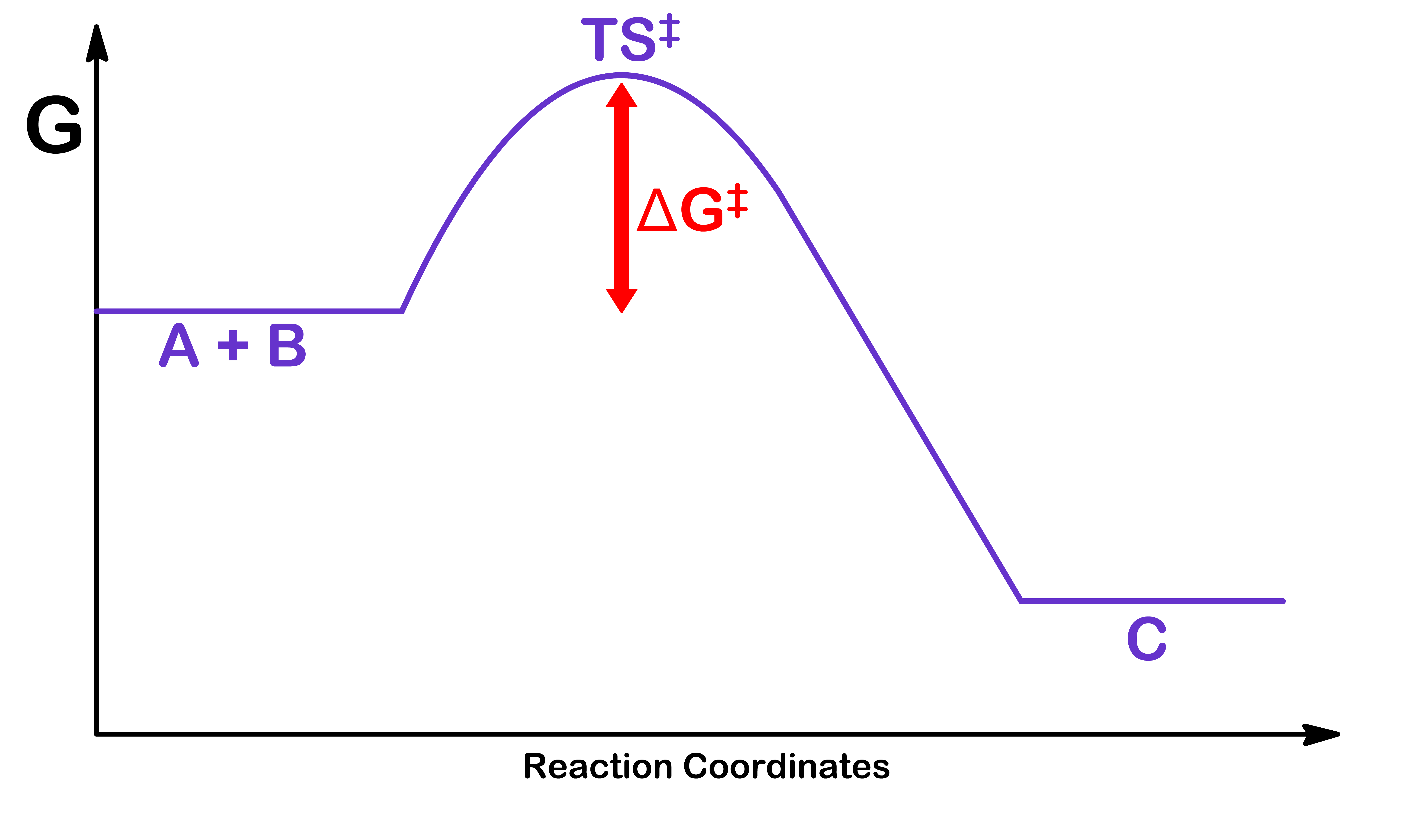
Let us first consider the generic reaction of A+B⟶C
Even though this is a one step reaction, there are three reactions taking place
The reactants collide to form the transition state
A+B⟶TS‡
The transition state breaks down and become the reactants again
TS‡⟶A+B
The transition state continues reacting and becomes the product
TS‡⟶C
Combining all three reactions, we can break down the one-step reaction into two different pieces
A+BK‡TSk‡C
The reactants are in a rapid pre-equilibrium with the transition state and the equilibrium constant is K^
The transition state reacts to form the product and the rate constant of this reaction is given by k^
Eyring-Polanyi equation
Having laid out the background for the transition-state theory, we can start deriving the Eyring-Polanyi equation
We are interested in the rate of reaction. Since this is a standard elementary reaction, we can write down the rate law as the product of each reactant's concentration raised to the power of 1 and the overall rate constant of the reaction
dtd[C]=k[A][B]
Since the rate is defined as the rate of change of the concentration of product, we can also think about the rate by only looking at the second portion of this reaction
dtd[C]=k‡[TS‡]
Equating the two expressions of rate gives us
k[A][B]=k‡[TS‡]
The concentration of the transition state is hard to measure. Fortunately, we can express it in terms of the concentration of the reactants
The Eyring-Polanyi equation has a shocking resemblance to the Arrhenius equation we learned in year 1
k=(κhkBTeRΔS∘−‡)e−RTΔH∘−‡k=Ae−RTEA
Simply by inspection, we can find an expression for the pre-exponential factor, A
A=κhkBTeRΔS∘−‡
We can therefore interpret A as the frequency of attempts to go over the transition state. Moreover, the inclusion of ΔS∘−‡ allows us to factor in the steric effects
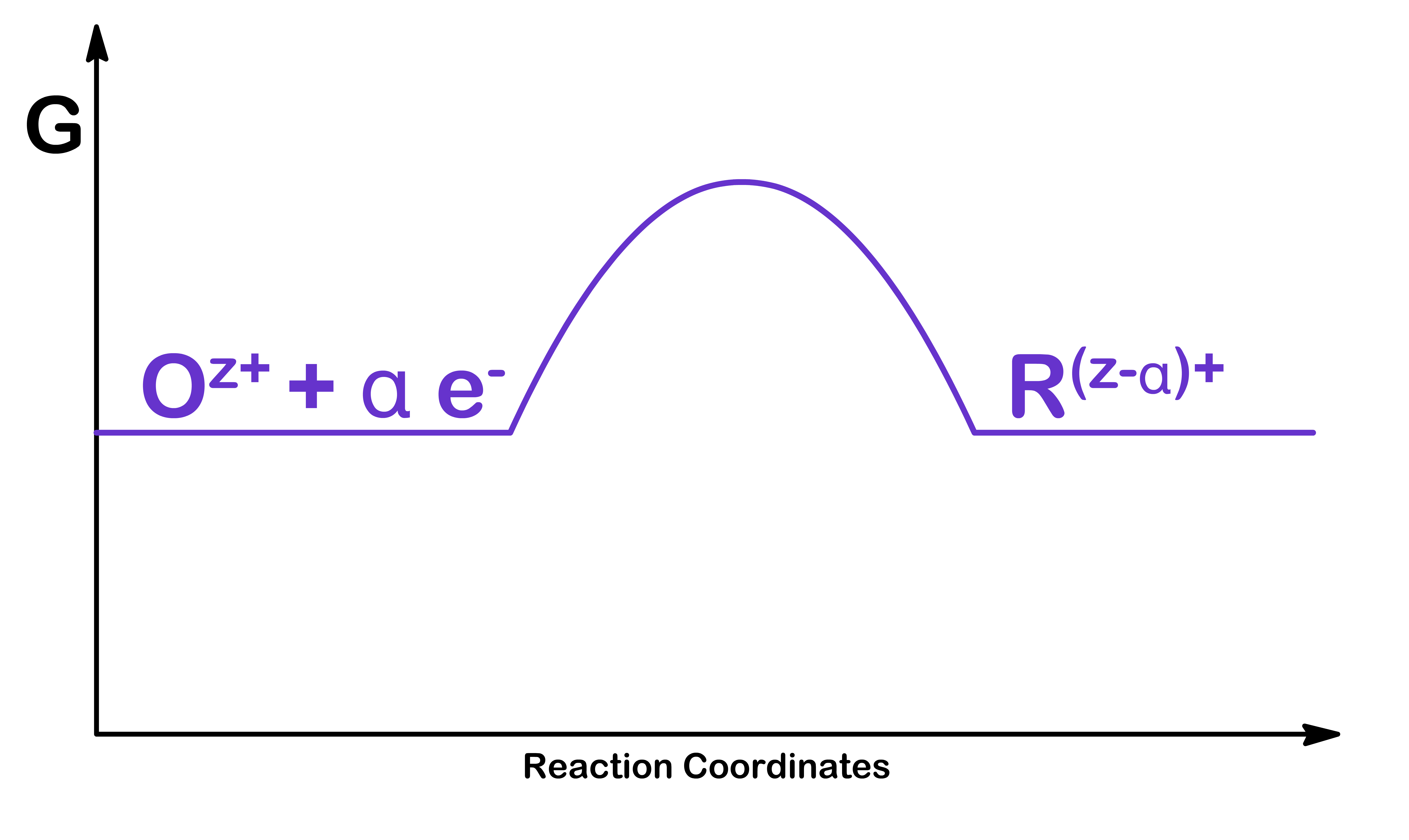
Let us consider the rate of the following generic half equation where we are assuming that both oxidation and reduction is taking place at the same electrode simultaneously
Oz++αe−R(z−α)+
In general, the rate of reaction can be characterized as the rate of change of reactants
rate=dtdN⋯⋯N=moles of Oz+
In electrochemistry, we can relate the charge carried with the moles of reactants consumed
dQ=αFdN⎩⎪⎪⎨⎪⎪⎧dQαFdNcharge transferredstochiometric coefficient of electronFaraday’s Constantnumber of moles of consumed reactants
Since the current, i , is defined as the rate of charge transferred, we shall divide both sides by dt
idtdQ=αFratedtdN⇓i=αF×rate⇓rate=αFi
To keep matters simple, we shall use rate per unit area as the parameter instead. Moreover, the current divided by the area will just be the current flux ( j )
Rate per areaArate=αF1×jAi⇓Rate per area=αFj
The overall rate is the sum of the forward and backward process
∵Oz++αe−kredkoxiR(z−α)+∴Rate per area=kred[Oz+]−koxi[R(z−α)+]⇓kred[Oz+]−koxi[R(z−α)+]=αFj
We therefore have an expression of the current flux
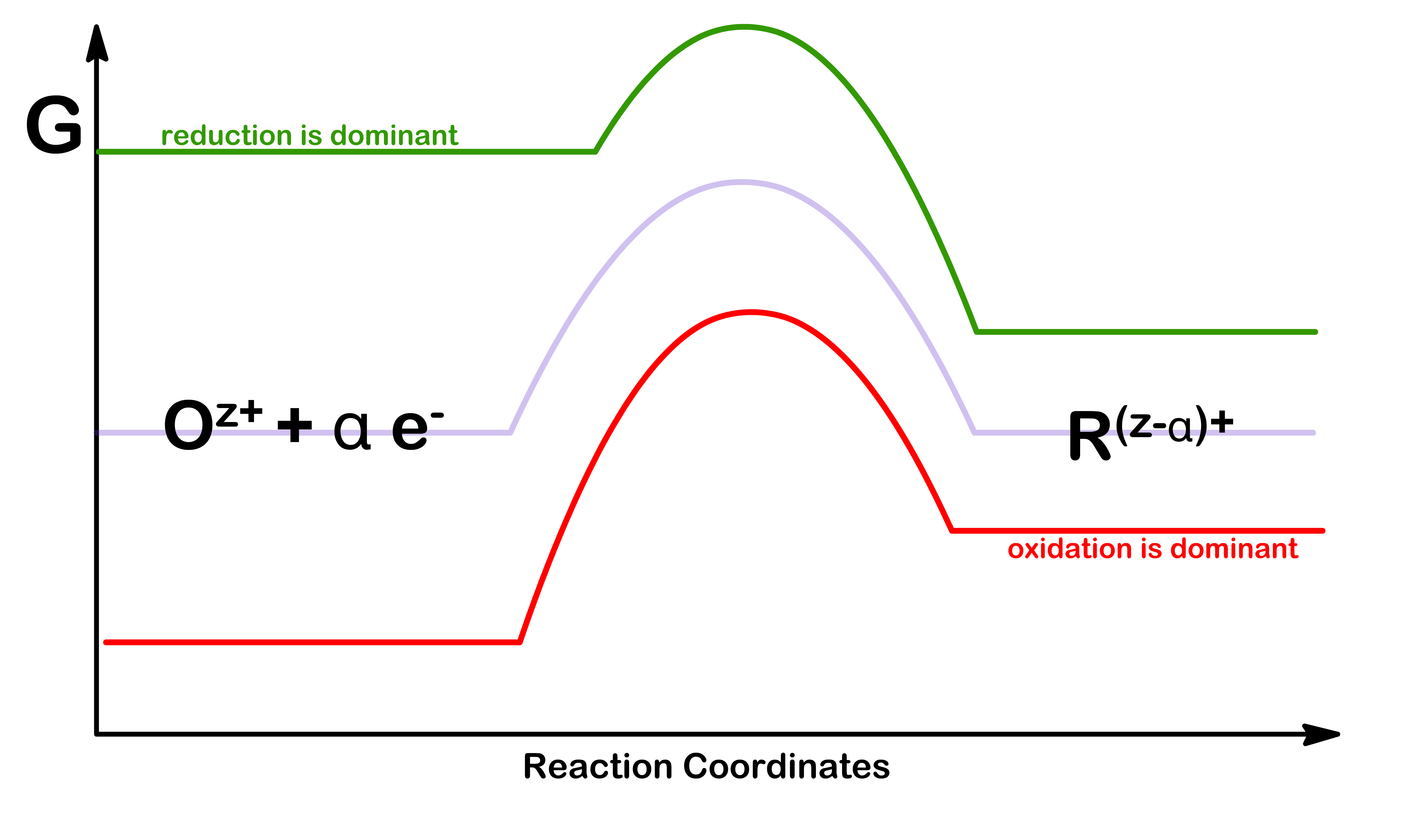
In electrochemistry, we can change the direction of a half reaction by by applying a potential at the electrode
Oz++αe−R(z−α)+
At equilibrium, the energy of the oxidized species and the reduced species should be the same. The potential at which this equilibrium establishes is denoted as Erev
By applying a potential, we can alter the energy of both the oxidized and reduced species. As a result, either oxidation or reduction will be dominant at that electrode
The overpotential ( η ) is defined to be the difference between the equilibrium potential and the actual potential at the electrode
η=E21cell−E21cellrev
The sign of the overpotential indicates the direction of the reaction at the electrode
The Butler-Volmer Equation can be derived from the equation we derived from the previous section
j=αF(kred[Oz+]−koxi[R(z−α)+])im too exhausted to make⇓ the derivation fun and intuitivej=j0(oxidationeRTαF(1−β)ηreduction−eRT−αFβη)Butler-Volmer Equation
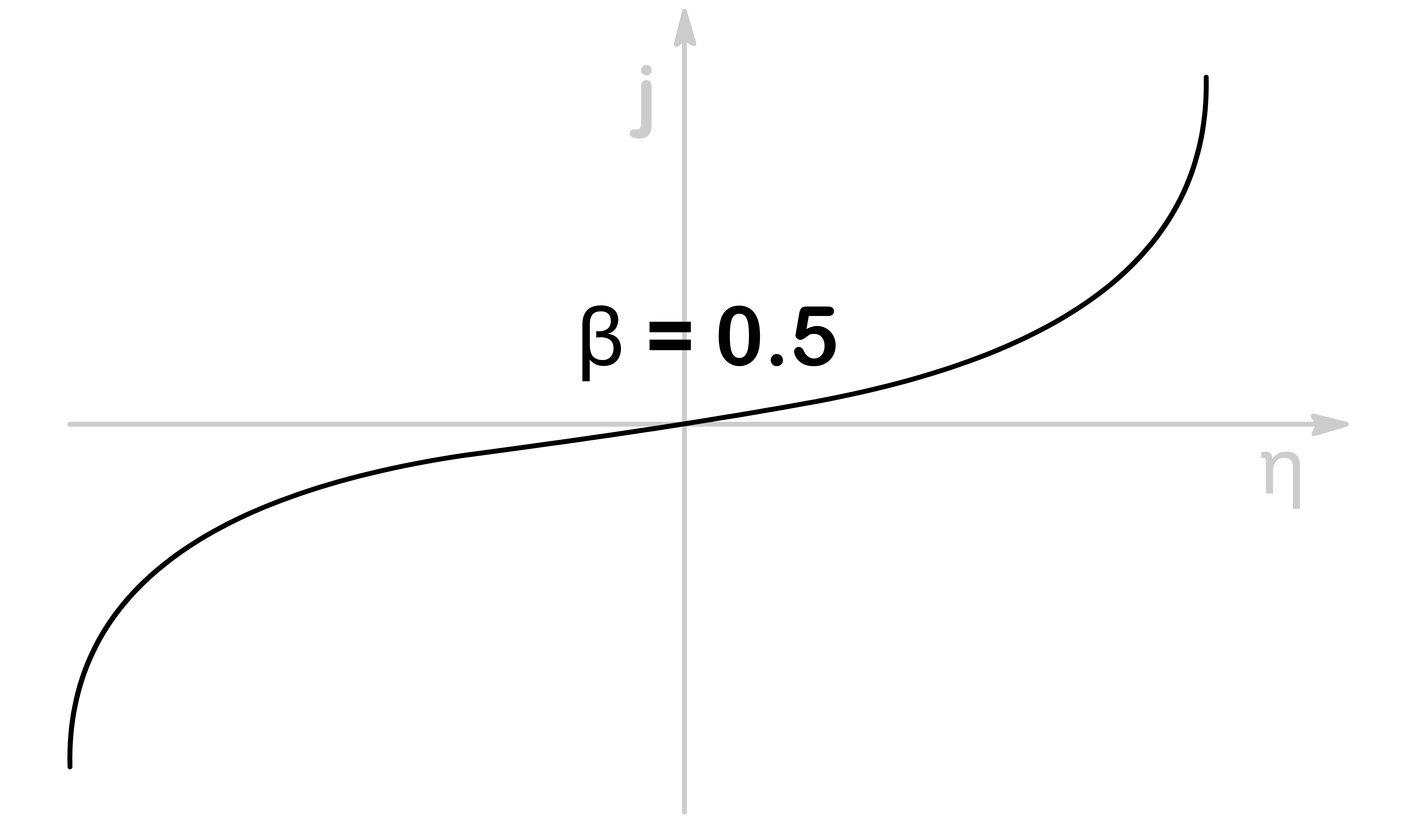
β is the symmetry factor and it is an indication of whether the transition state is reactant-like ( β=0 ) or product-like ( β=1 ) or somewhere in between ( β=21 )
j0 is the exchange-current density. It is the magnitude of the equal but opposite current density when the electrode is at equilibrium
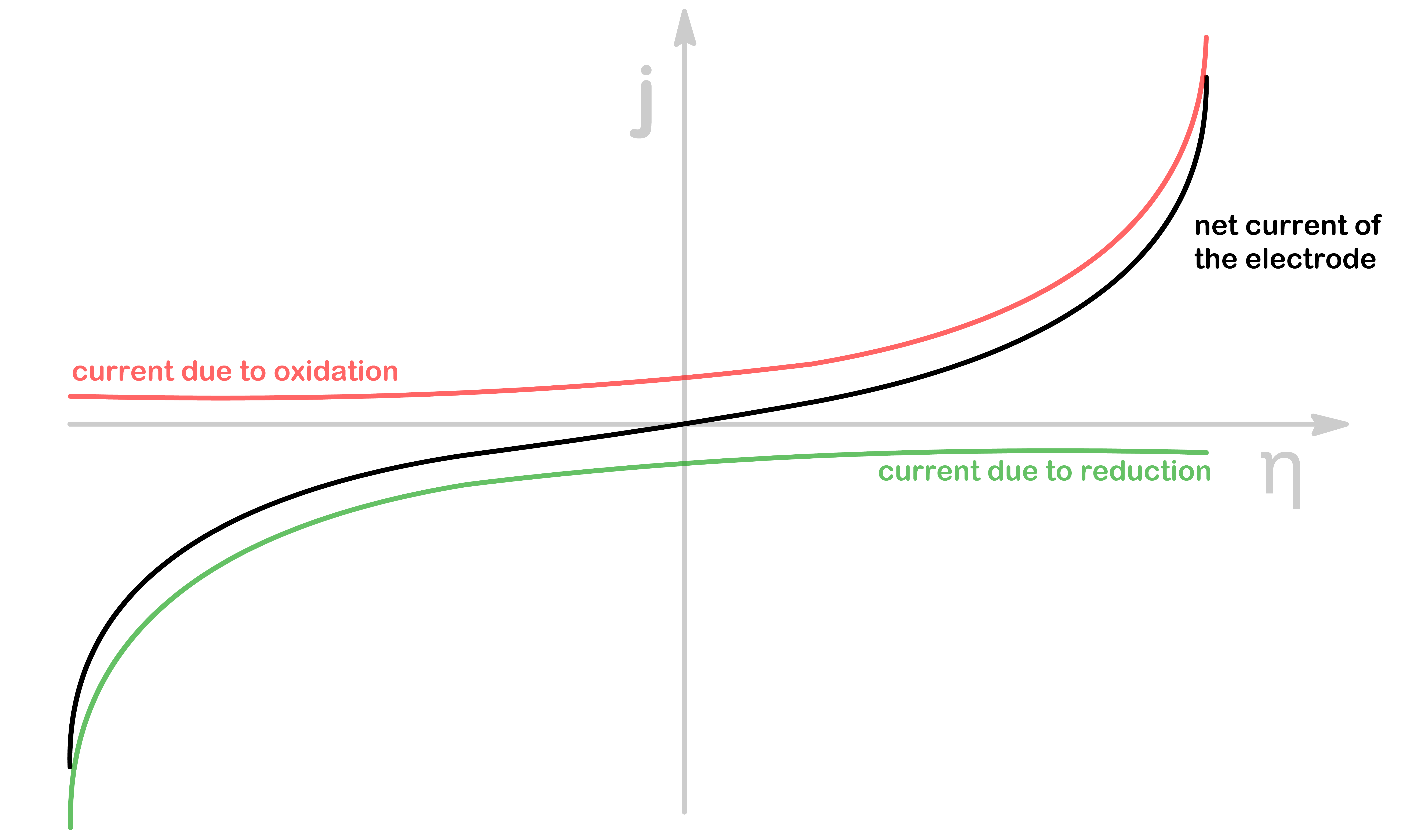
The Butler-Volmer equation gives us the same conclusion as above as the the current flux of one electrode changes with η
When the overpotential is positive, the current will also be positive ( η>0→j>0 ). The electrode will therefore be the anode
When the overpotential is negative, the current will also be negative ( η<0→j<0 ). The electrode will therefore be the cathohde
Limiting forms and the Tafel Plot
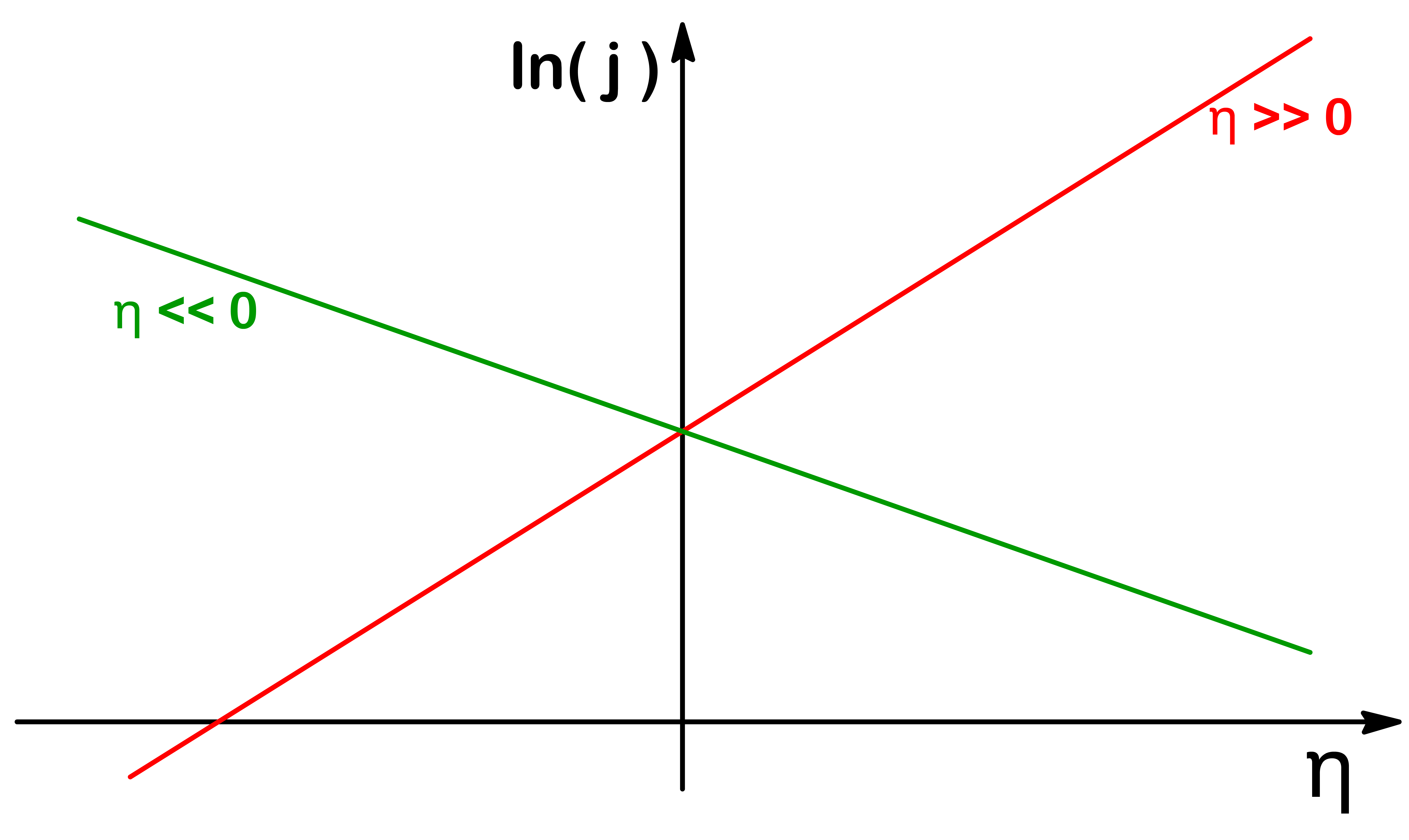
As shown in the above plot, when the magnitude of η is large, the net current of the electrode will be approximately the current due to oxidation or the current due to reduction
In these situations, one of the exponential is much bigger than the other, so we can omit that term
When η>>0:j=j0(eRTαF(1−β)η−eRT−αFβη)⇓j≈j0(eRTαF(1−β)η)When η<<0:j=j0(eRTαF(1−β)η−eRT−αFβη)⇓j≈j0(−eRT−αFβη)
The simplied forms of the Butler-Volmer equation allow us to plot a linear graph
When η>>0:j≈j0(eRTαF(1−β)η)⇓yln(j)=mRTαF(1−β)xη+cln(j0)When η<<0:j≈j0(eRT−αFβη)⇓yln(j)=mRT−αFβxη+cln(j0)
By determining the slope and the y-intercept, we can find out the value of β and j0 respectively
More on j0 and β
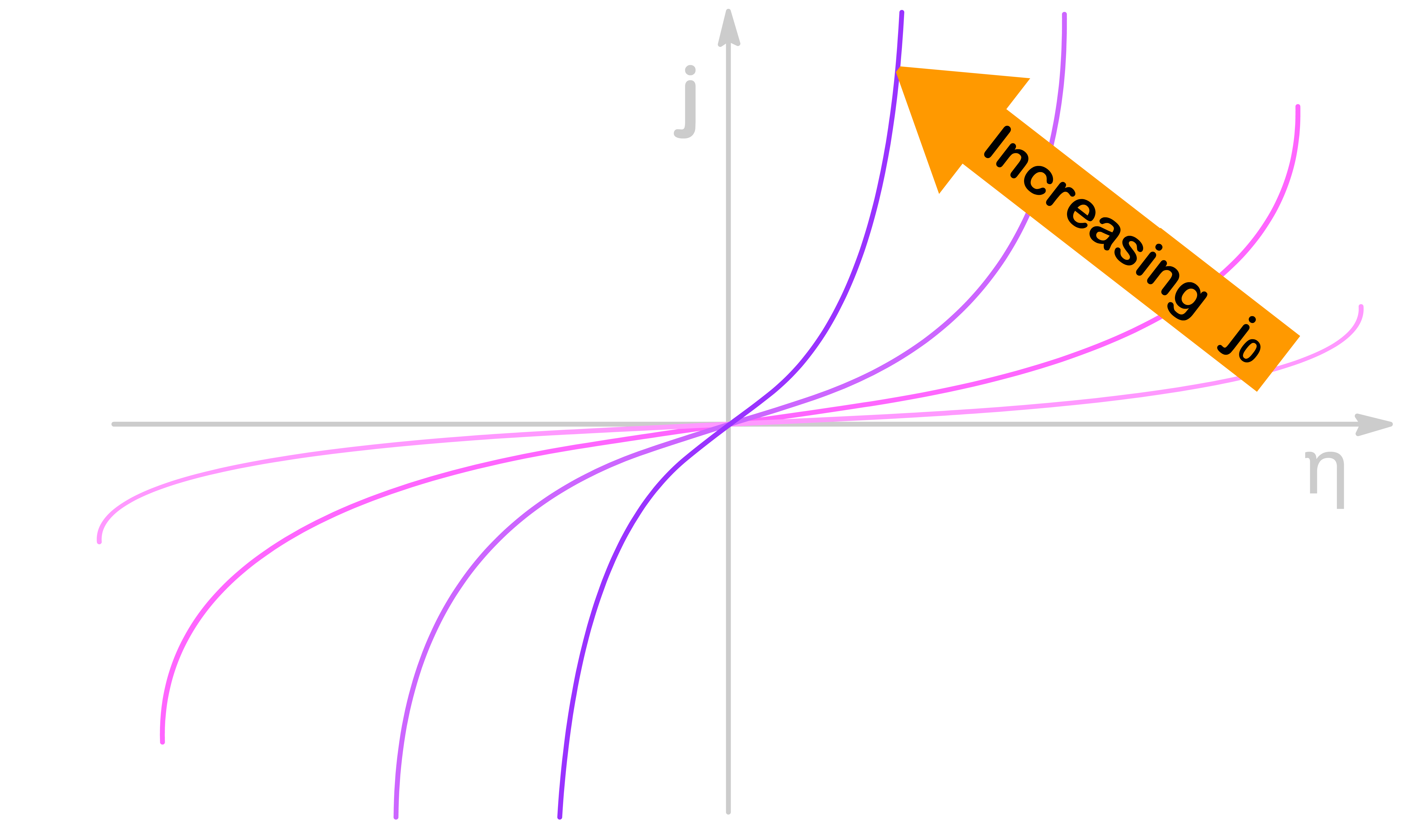
j0 can be thought of as a measurement of how easily the reaction occurs
A large j0 is associated with a reaction that can be accelerated very quickly with a relatively low overpotential
As established before, the y-intercept of the logarithmic graph is given by ln(j0), so an increasing value of j0 will shift the graph upward
If we plot j0 against T1, we will get a straight line graph where the slope is −RΔ‡Hrev
β represents the degree to which the transition state is similar to the reactant or product
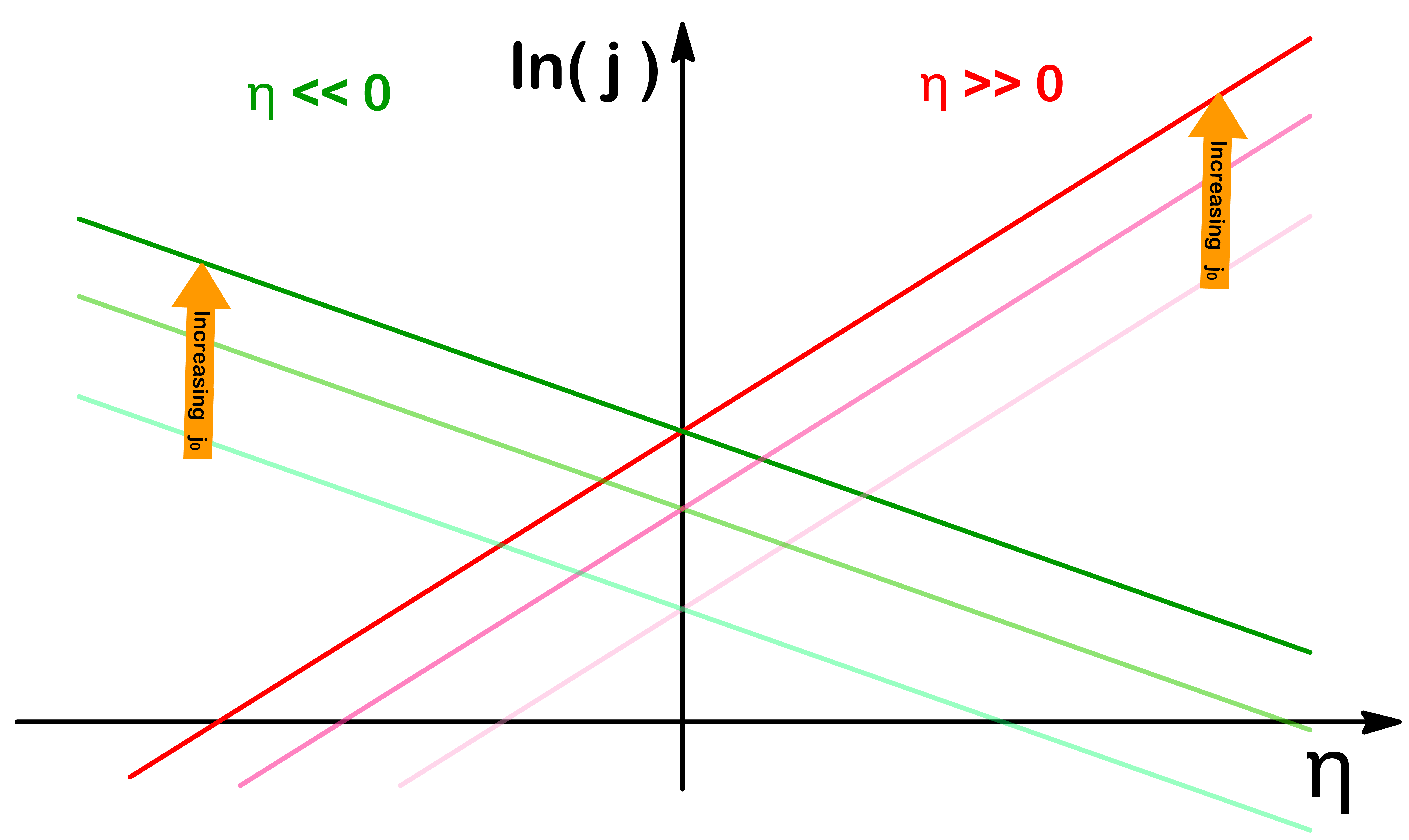
When β=0.5, the response towards the applied potential is symmetrical for both oxidation and reduction
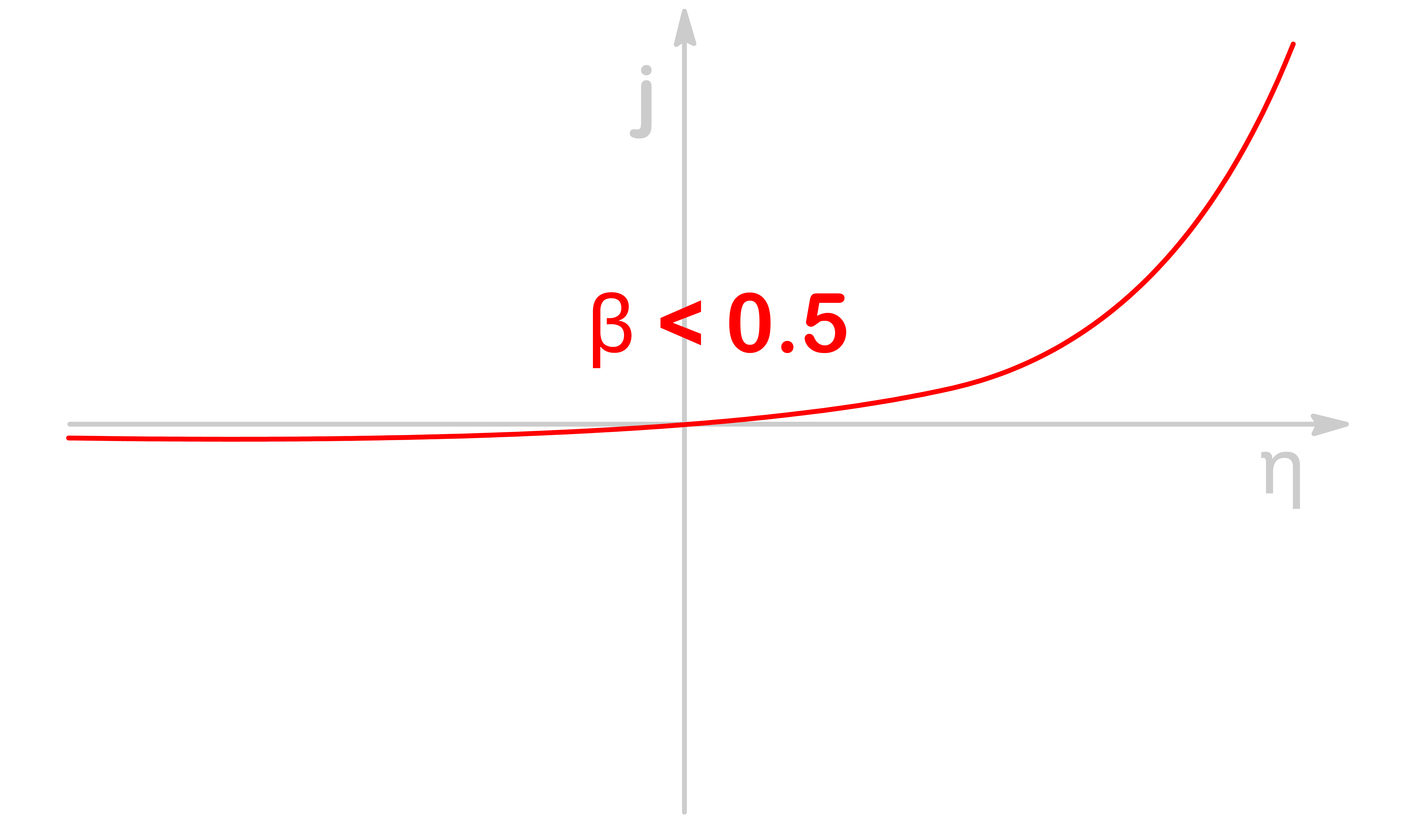
When β<0.5, the transition state is more similar to the oxidized spiecies, so the rate of oxidation accelerates much faster than the rate of reduction for a given amount of overpotential
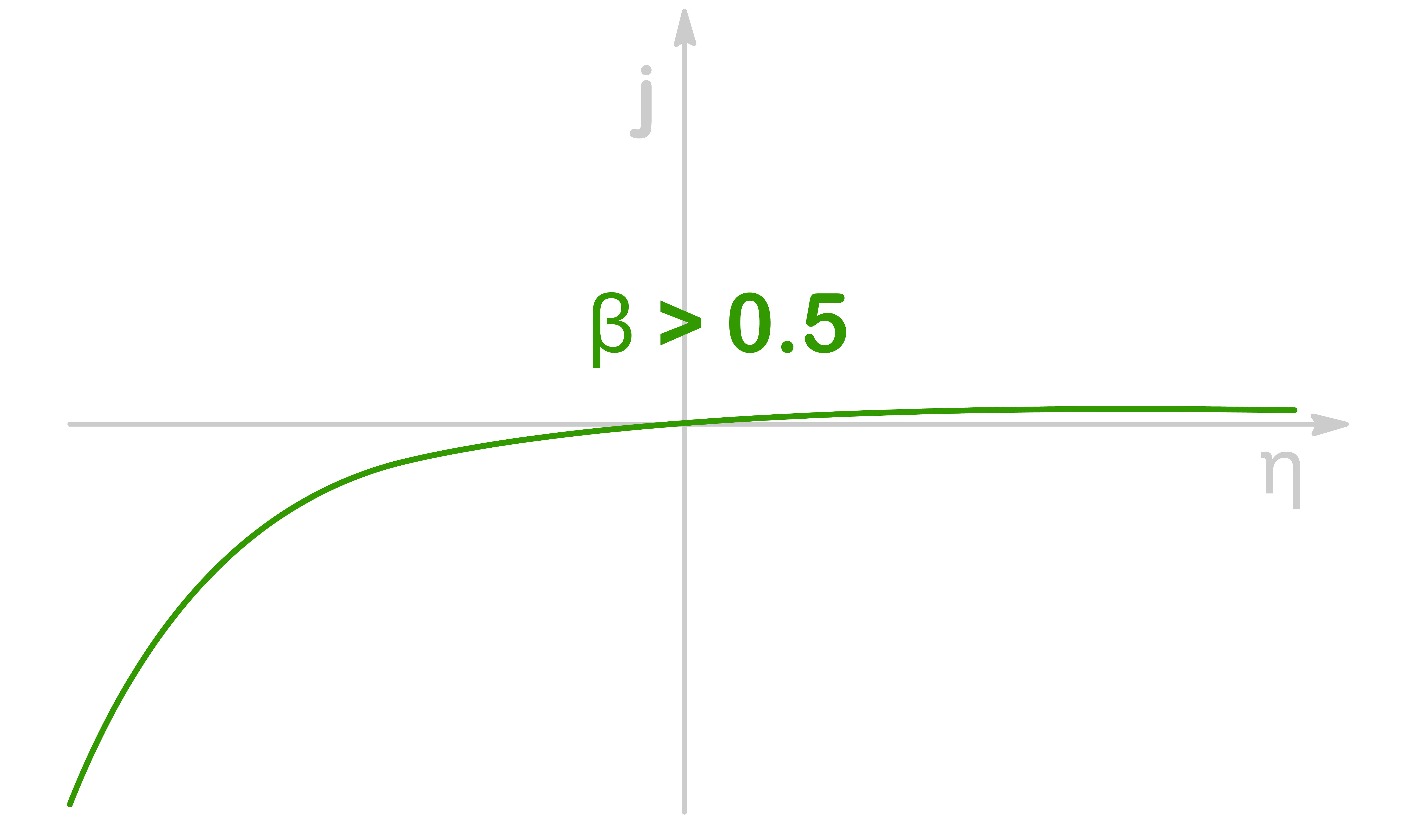
When β>0.5, the transition state is more similar to the reduced spiecies, so the rate of reduction accelerates much faster than the rate of oxidation for a given amount of overpotential
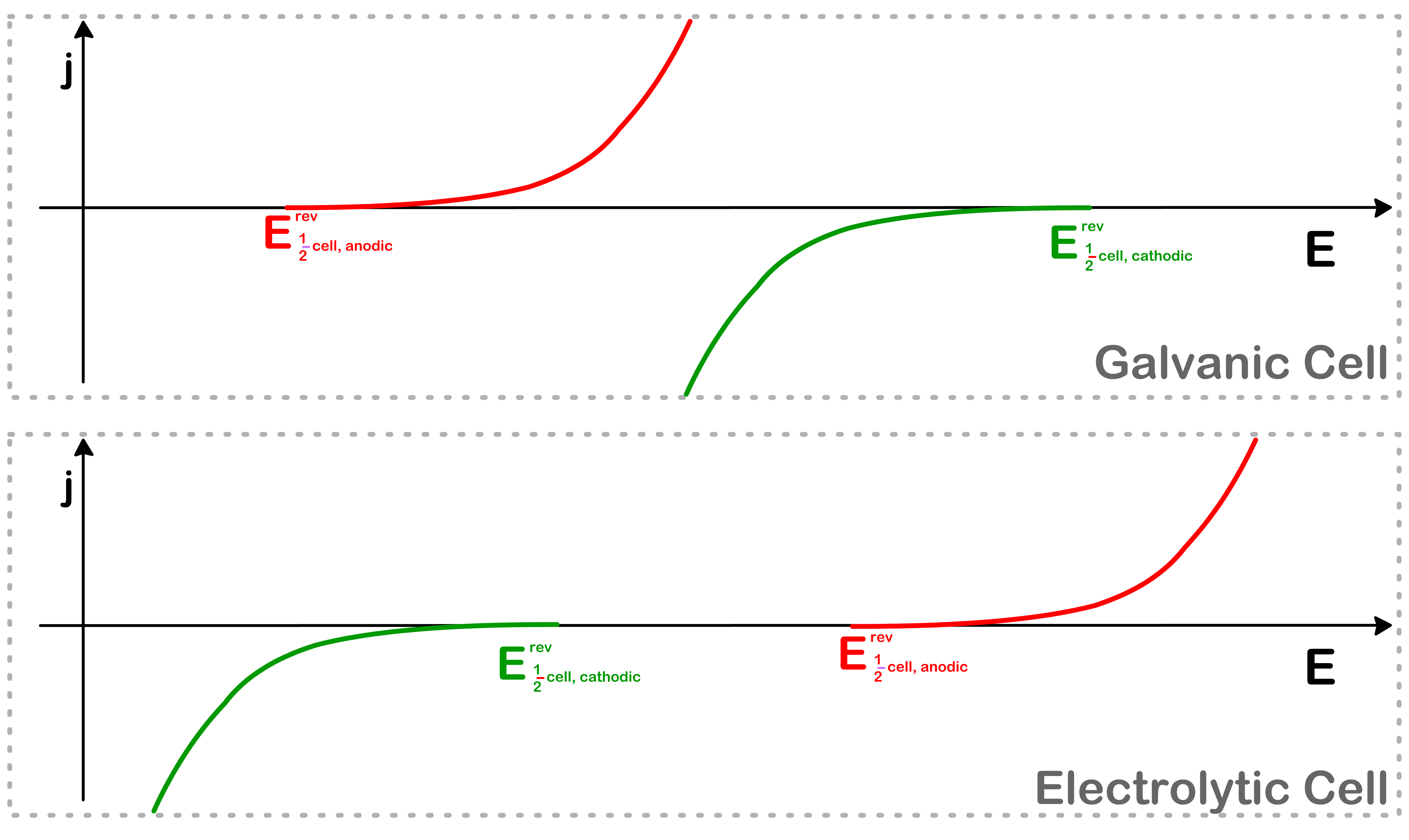
In a full-cell, the current flux ( j ) of the cell can only have one value at any given moment, but there can be two values of η
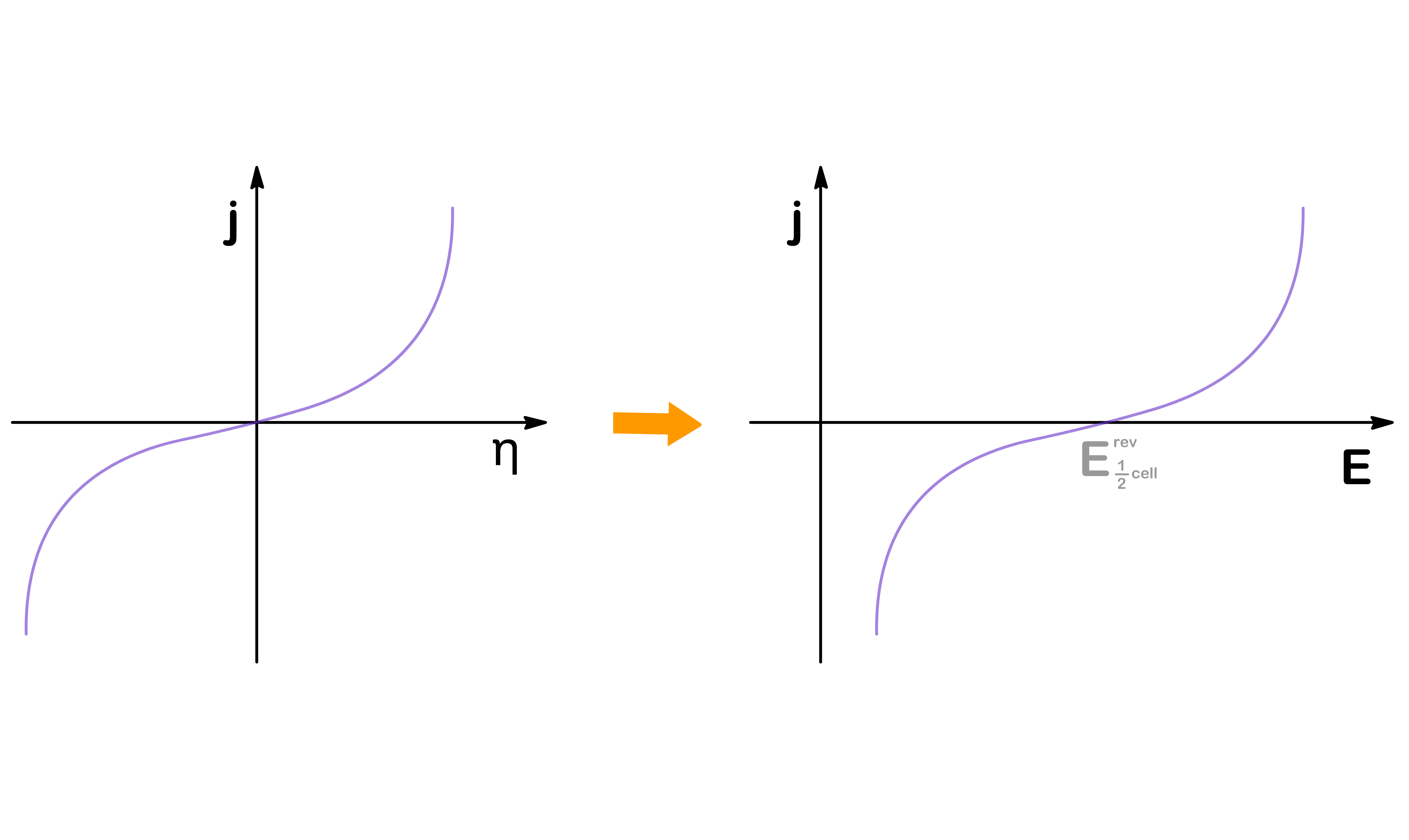
We therefore wish to change the x-axis from η to E. This can be achieved by acknowledging that η=E21cell−E21cellrev
The shape of the graph will remain the same, but the x-intercept will go from η=0 to E=E21cellrev
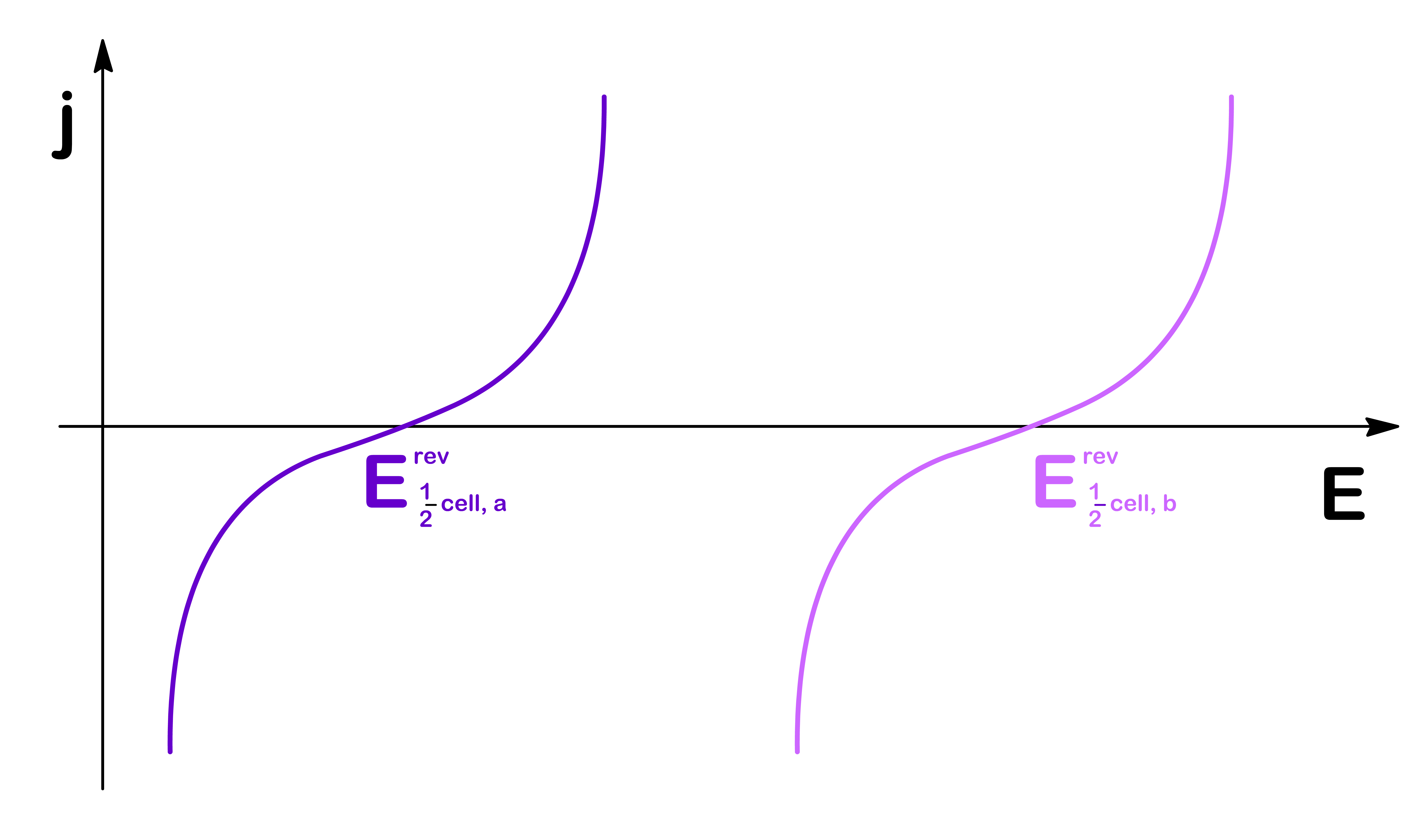
We can now combine the two half-cells and plot them on the same graph
Depending on whether the cell is galvanic or electrolytic, the shape of the graph will look different
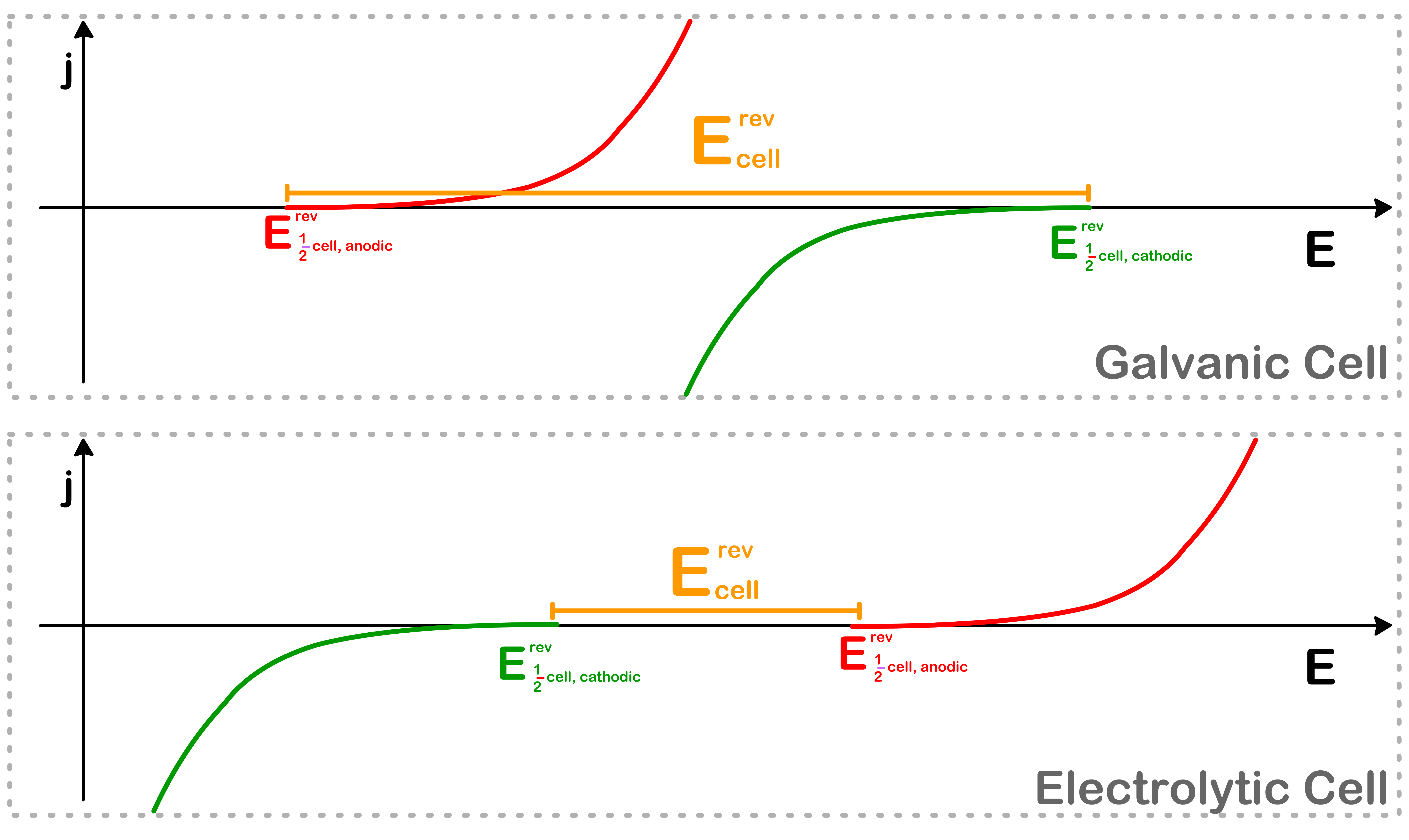
It is tempting to think that the cell potential is just given by the difference of the equilibrium half-cell potential. However, we will only be able to get the full-cell potential of a system that is in electroequilibrium
When the cell is operating, a current is passing through the cell, which means it is not in electroequilibrium and Ecellrev will not be the cell potential in this situation
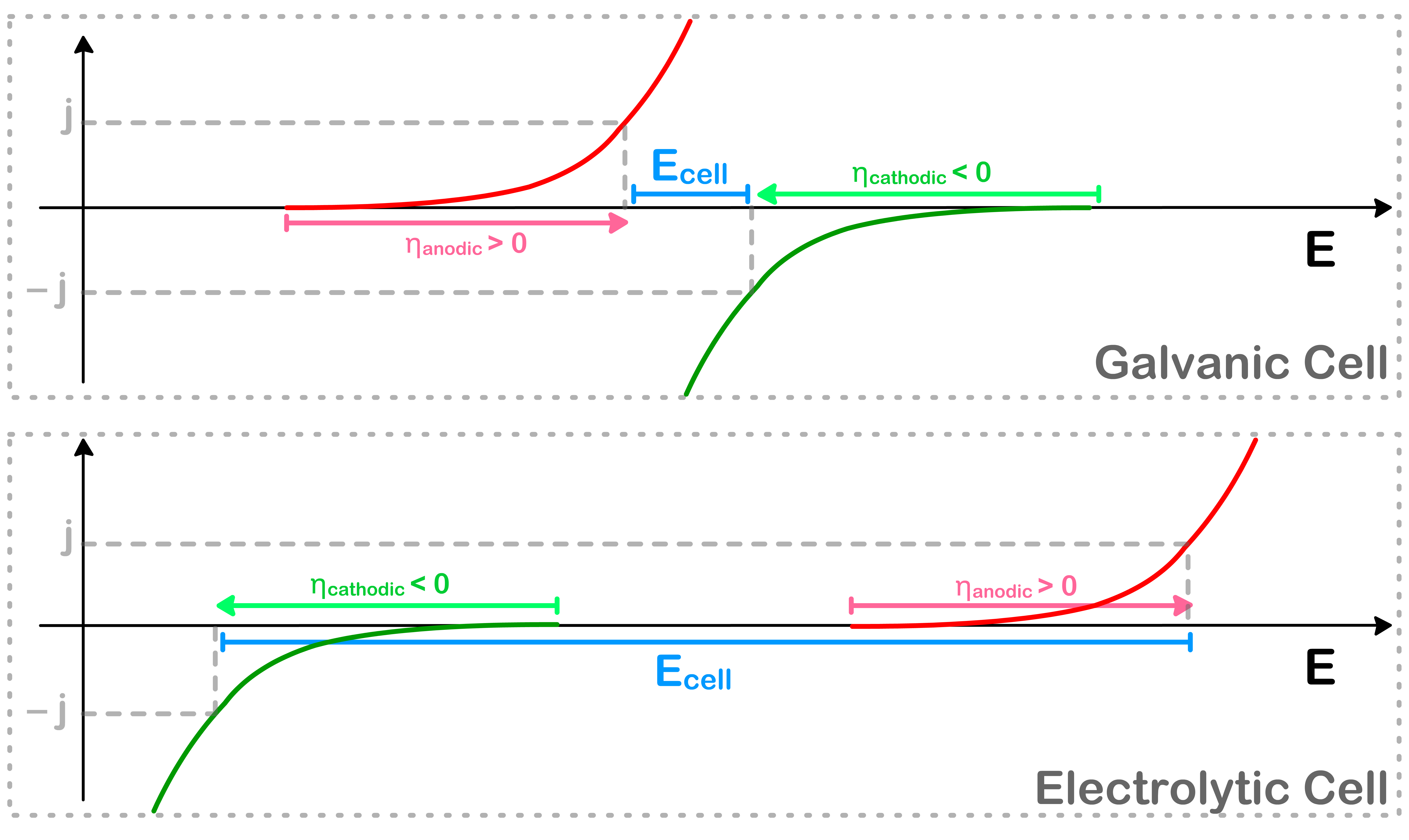
Suppose the cell is running with a current flux of magnitude j. The corresponding cell potential can be found by determining the potential at j and −j respectively
We will also be able to identify the corresponding overpotential for each half-cell using this method
¶ Time dependence of transport of species to interfaces
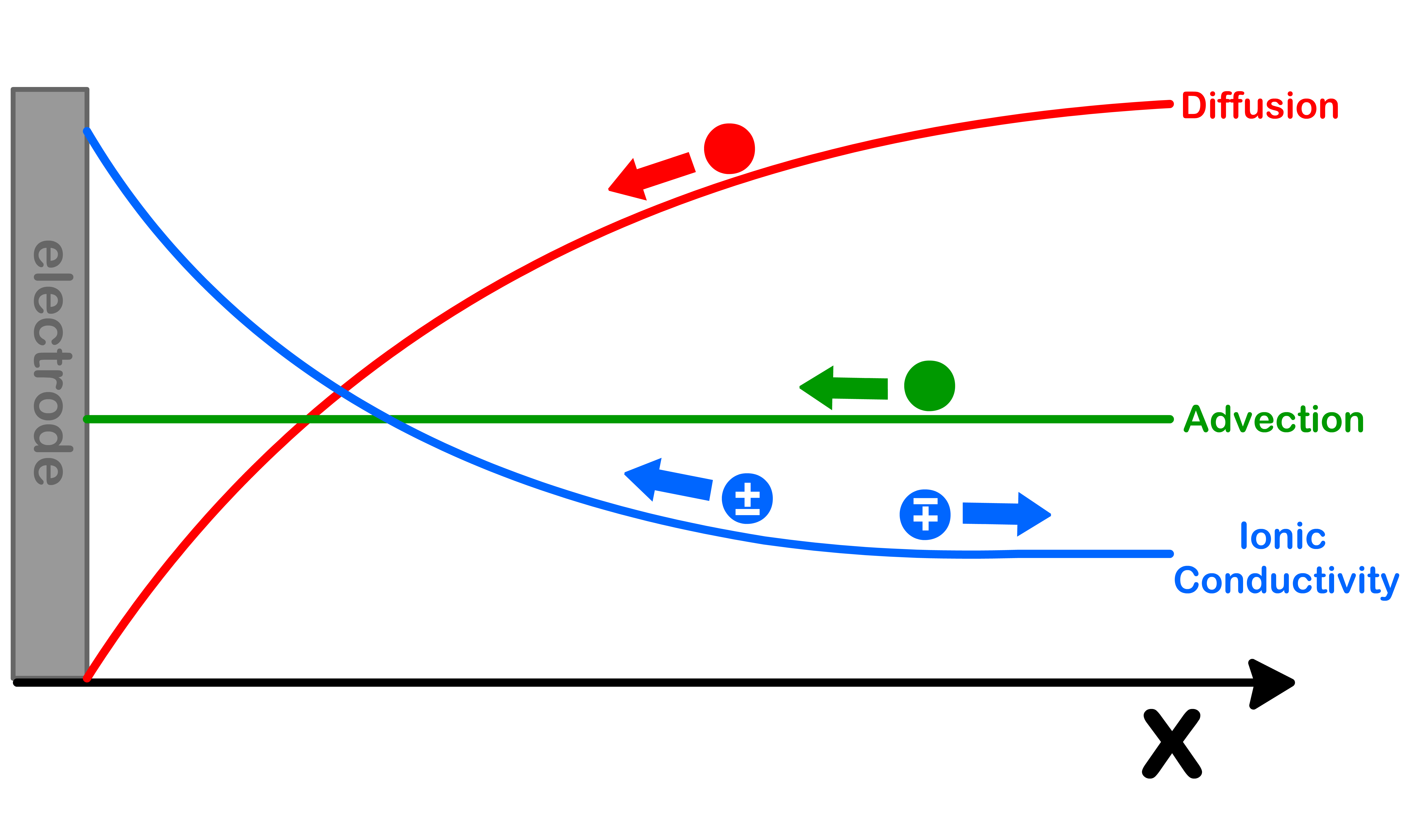
In dilute solution the time dependent flux ( J ) of ionic species transported to a solid surface in one dimension is described by the 1-D Nernst-Planck equation
J(x,t)flux to surface=diffusion−D(∂x∂c)−RTzFDc∂x∂ϕsionic conductivityadvection+vxc
The Nernst-Planck equation discusses the flux to the surface of the electrode as a function of time and space, and it is dependent on three components
Rate Limiting Step
When the reaction is taking place on an interface, the concentration of the reactant on the surface will deplete. We therefore have to transport the reactant from the bulk.
In effect, we can define three domains depending on the ratio of the rate of mass transport and the rate of reaction
kRxnkMT>10⋯⋯kinetic limited reactionkRxnkMT<0.1⋯⋯mass transport limited reaction0.1<kRxnkMT<10⋯⋯mixed regime
Since we are dealing with electrochemistry, instead of using the ratio of the rate constants, we can instead use the ratio of the current flux
We shall consider two scenarios
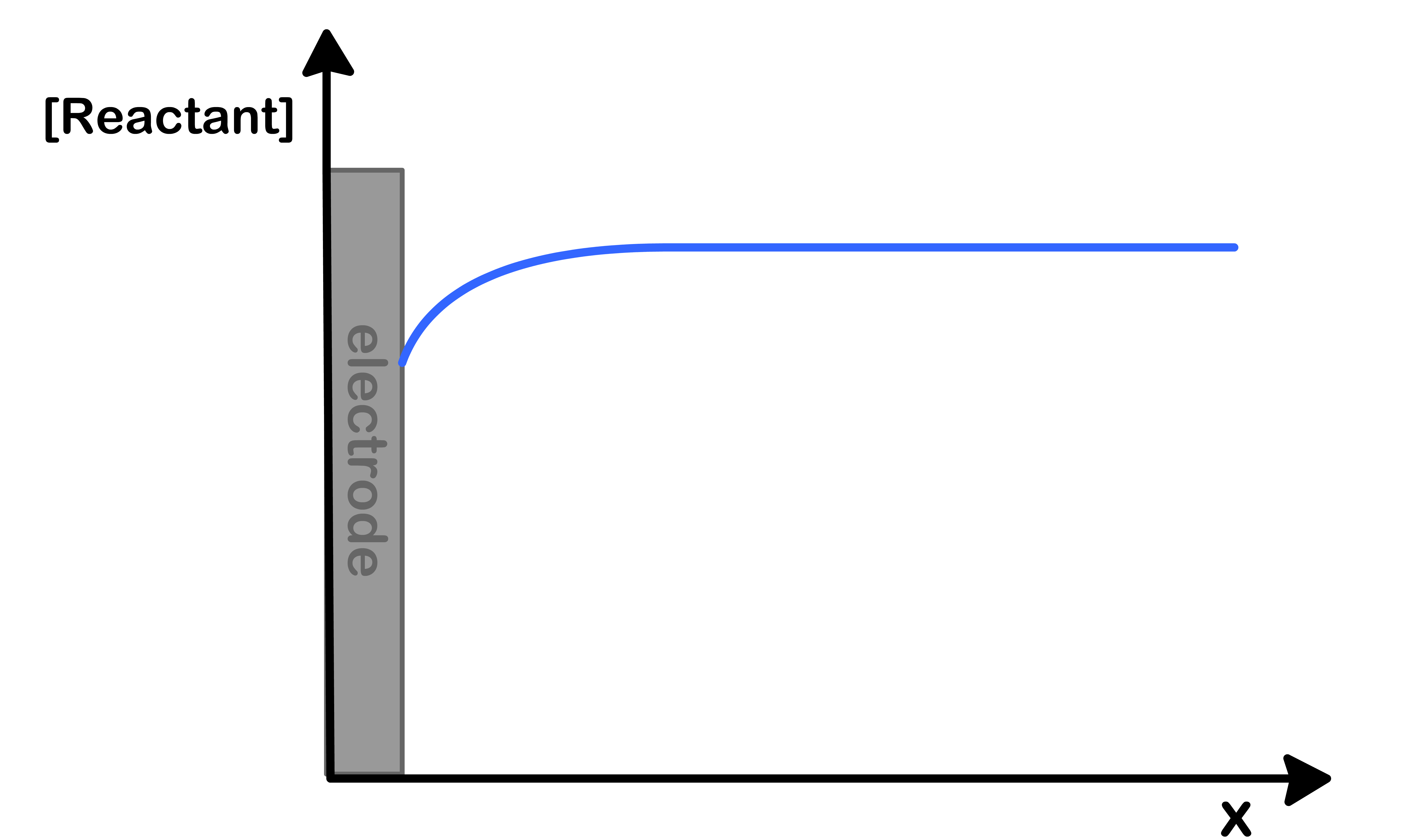
Case 1: When the electrochemical reaction is kinetic-limited
Case 2: When the electrochemical reaction is mass-transport-limited
Case 1: Kinetic Limited Reactions
jRxnjMT>10
This happens when the diffusion of reactants to the electrode is fast but the reaction itself is slow
This means that the reactants are replenished to the surface before they can be consumed. As a result, the concentration of reactant at the surface is time-independent
Moreover, the current is also time-independent apart from initial capacitance and slight perturbation of concentration at surface
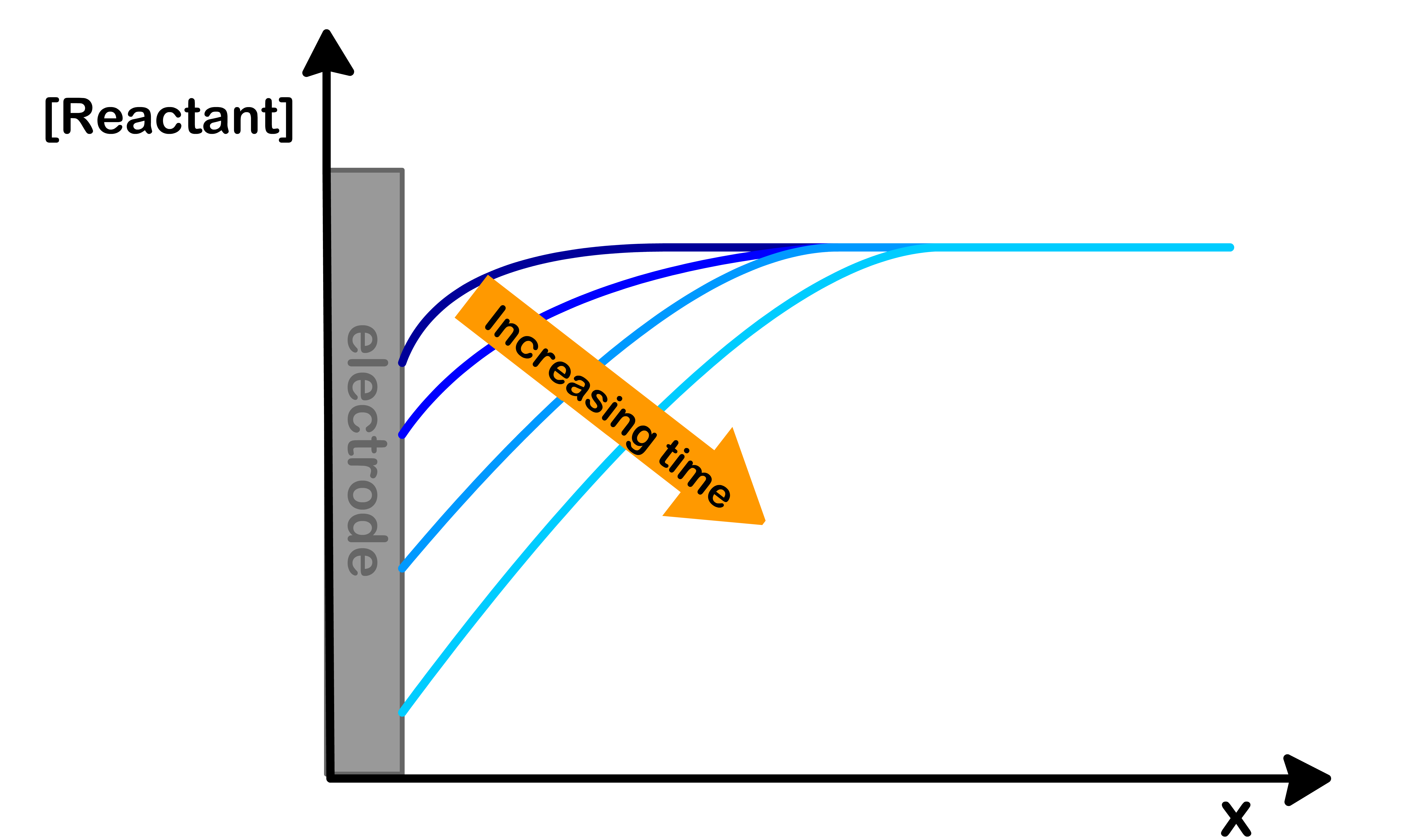
Case 2: Mass Transport Limited Reactions
jRxnjMT<0.1
This happens when the diffusion of reactants to the electrode is slow but the reaction itself is fast
This means that the reactants at the surface are consumed before they can be replenished. As a result, the concentration of reactant at the surface is time-dependent
Moreover, the current flux is also time-dependent and is given by the Cottrell equation
We shall now explore a technique that allows us to make the mass transport rate constant, kMT , to be independent of time
kMT=δDDOz+
For mass transport limited reactions, the diffusion distance ( δD ) is time-dependent, which in effect makes the corresponding kMT time-dependent as well
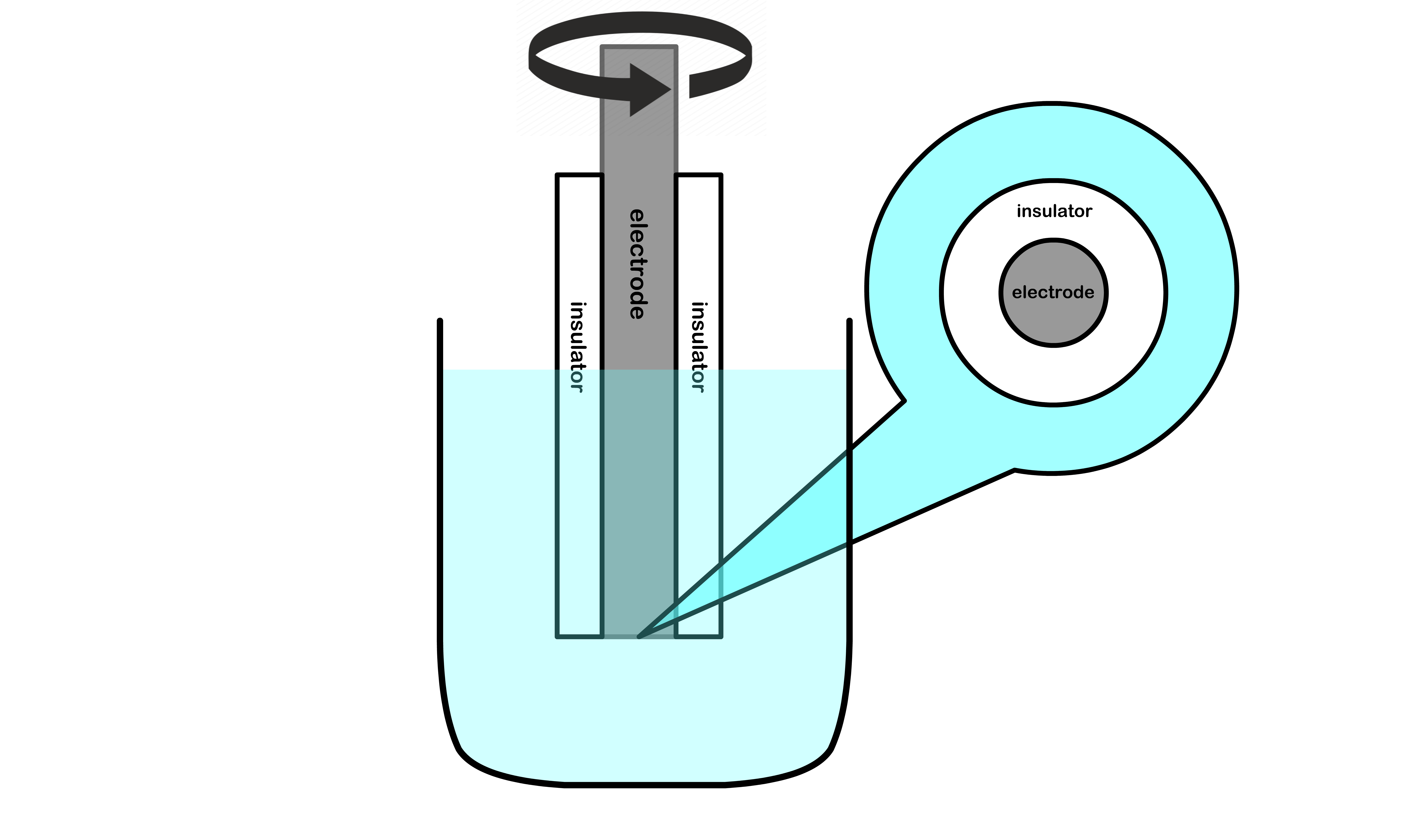
What we can do is to is to make use of a rotating disk electrode such that δD is fixed in time. As a result, the kMT of mass transport limited reactions can also become time-independent
Theory
The rotating electrode is embedded within an insulator such that electrochemical reaction can only take place at the bottom of the electrode
Due to the rotation, the flow near the electrode will be almost parallel to it
Base on the rotating speed, we can determine the current flux using the Levich equation