¶ Reactivity of Carbonyl Derivatives
Carbonyls almost always behave as electrophiles due to the polarity of the C=O bond
- Within the carbonyl functional group, the electrons in the σ and π bonds are drawn towards the more electronegative oxygen atom
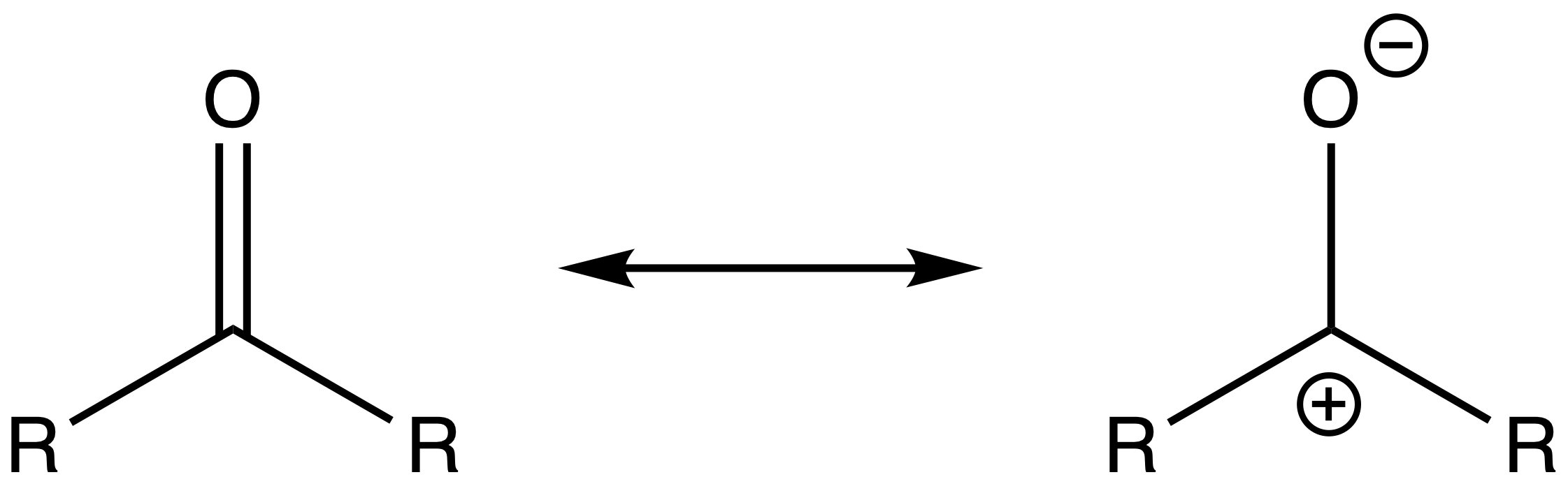
- In addition, the resonance structure emphasizes the reactivity of the carbonyl carbon as a electrophile and the carbonyl carbon as a nucleophile
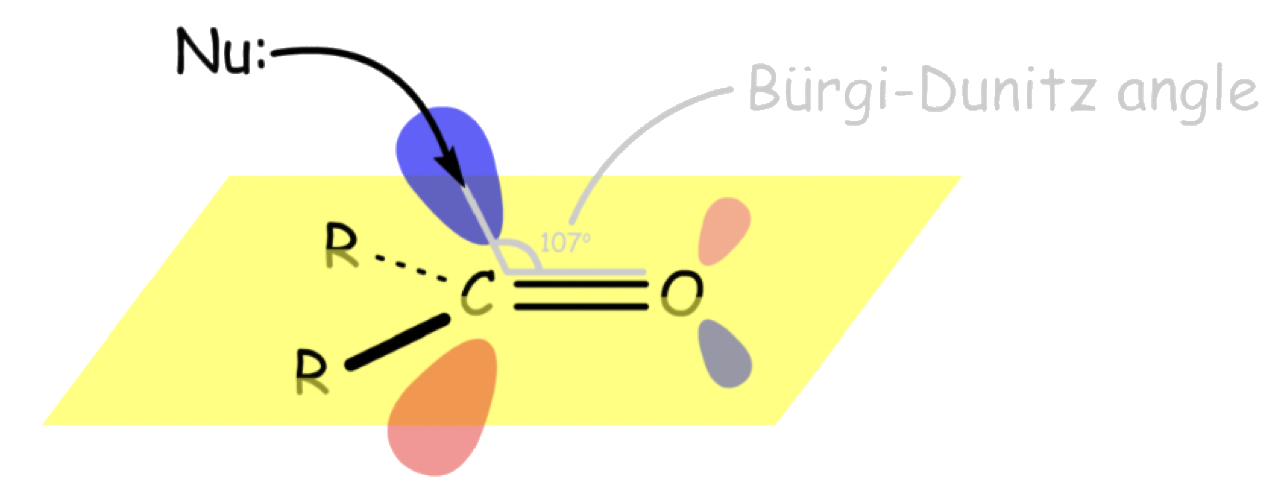
The nucleophile will attack the carbon through the π* orbital
- The nucleophile attacks at about 107° relative to the plane ( ∠O C Nu ) of the molecule. This angle is called the Bürgi-Dunitz angle
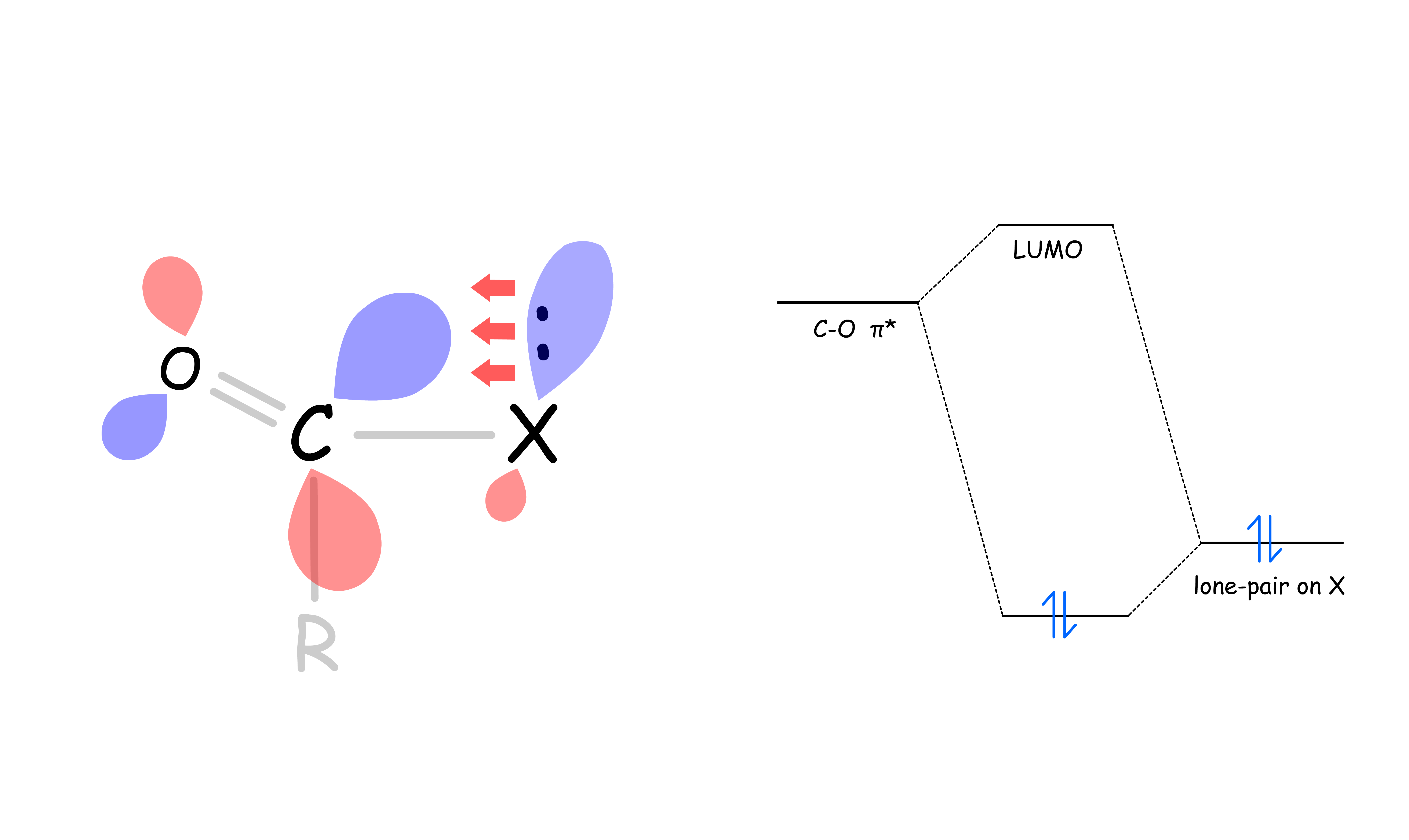
Not all carbonyls are created equally, some are more reactive than others (more electrophilic)
- The electron of the substituent in the σ orbital is donating electron density towards the empty C–O π* orbital. This makes the C–O π* orbital higher in energy, thus less electrophilic
- The effect becomes less prominent as the electronegativity of X increases, so the decrease in reactivity is not as apparent as the electronegativity of X increases
- In other words, the reactivity of carbonyls can be ranked as followed
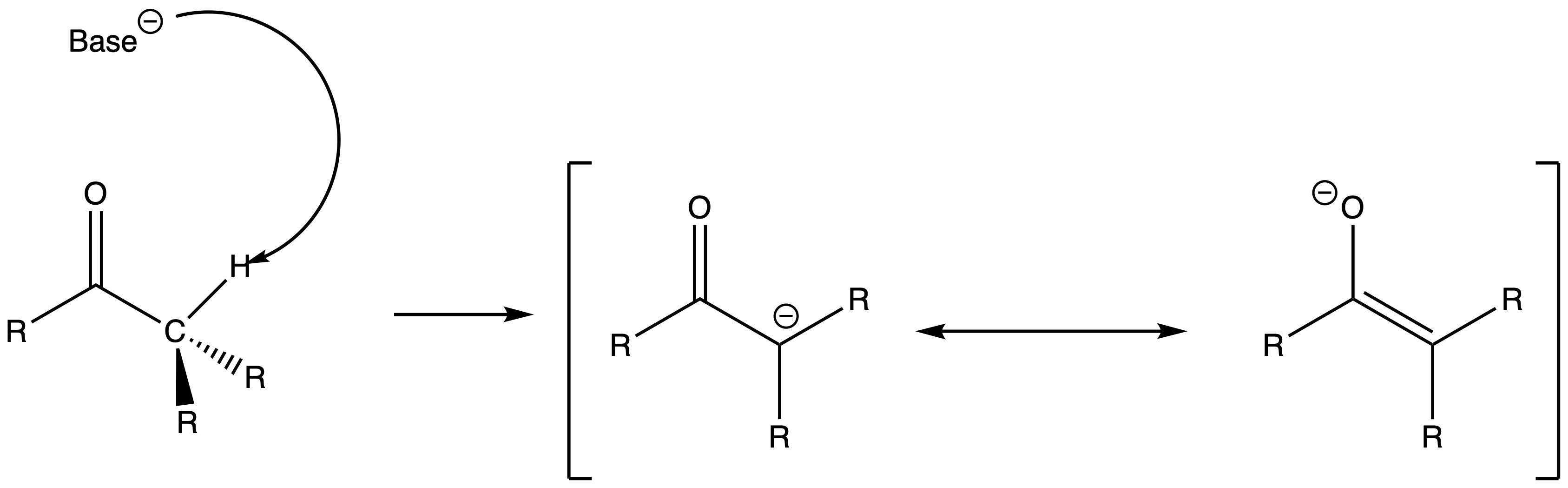
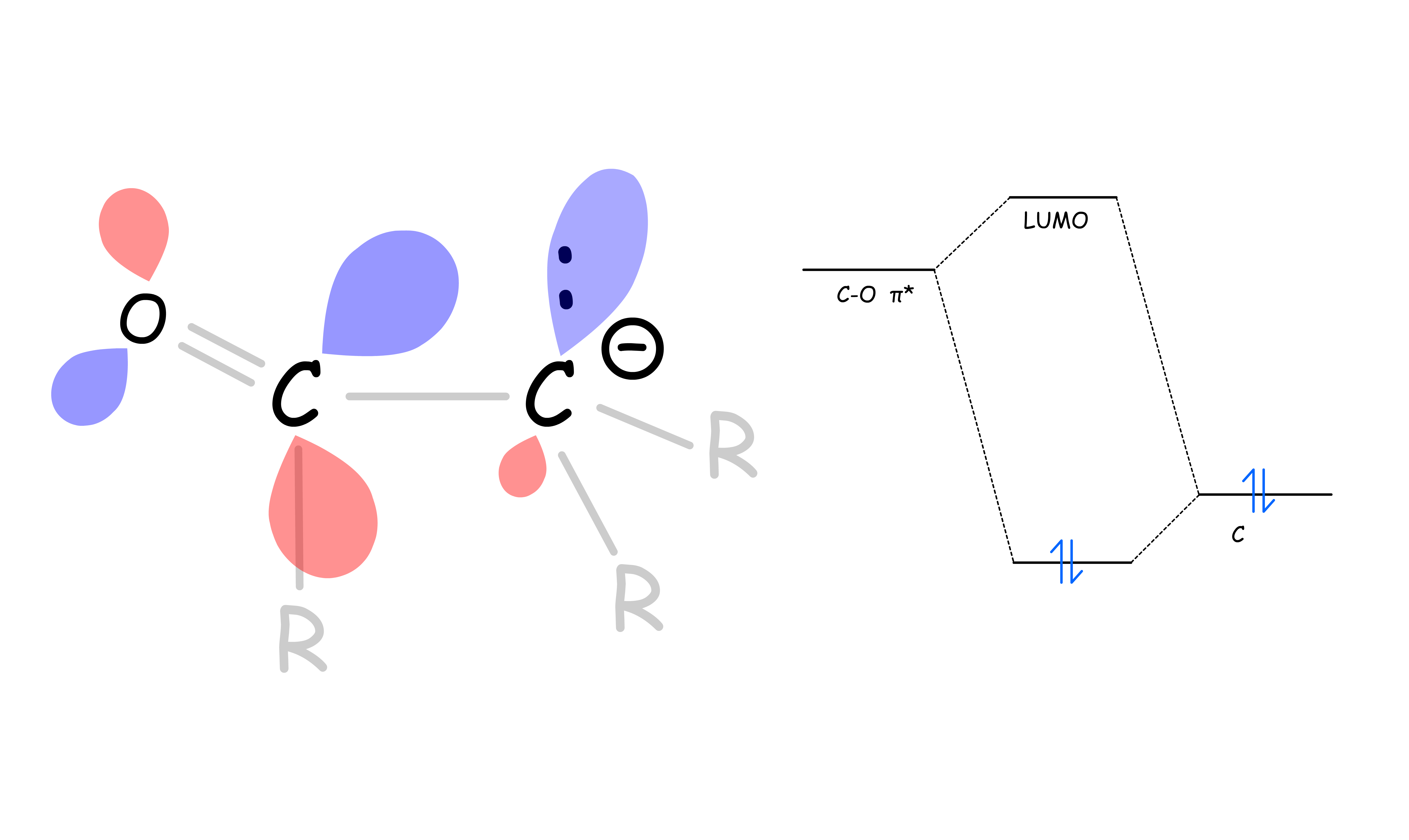
The alpha hydrogens of carbonyls are acidic
- α hydrogens are weakly acidic because the conjugate base, called an enolate, is stabilized though conjugation with the π orbitals of the carbonyl
- From a molecular orbital perspective, the orbital mixing between the filled sp3 orbital and the empty C–O π* orbital of the conjugate base allows the electron pair to be pulled deeper in energy level, thus more stable
- It is worth noting that if the nucleophilic attack is hindered enough, the carbonyl compounds may take part in an acid-base reaction instead
¶ Selectivity
¶ Chemoselectivity
The most reactive carbonyl wins
- This only works if the carbonyl you want to react with is the most reactive one by a significant margin
- This approach does not allow the use of exccessive amount of nucleophile as it will start to attack the other sites once the most reactive site is exhausted
Protecting ketone and aldehyde from reactions
- We can first convert the aldehyde or ketone to acetal first
- We can then let the other group do its thing
- We then convert the acetal back to the aldehyde or ketone
Halt the reaction at the tetrahedral intermediate using the Weinreb amide
- We first convert our carbonyyl to the Weinreb amide
- We then use organometallic reagent to react with the Weinreb amide
- The coordination stabilizes the tetrahedral intermediate and prevent it from collapsing. As a result, the carbonyl is not reformed and only one nucleophile is added
- Subsequent treatment with aqueous acid as a work-up liberates the ketone via a hemiaminal
A stable tetrahedral intermediate can be obtained using an excess of an organolithium with a carboxylic acid
- The first organolithium will behave as base and deprotonate the carboxylic acid
- The second organolithium will behave as a nucleophile and attack the carbonyl carbon
- The tetrahedral intermediate (dilithio dianion) formed does not have any good leaving group, so it will not collapse. As a result, the carbonyl is not reformed and only one nucleophile is added
- Subsequent treatment with aqueous acid as a work-up gives hydrate, which collapse back to a ketone
- note that a Grignard reagent is NOT a good enough nucleophile to do this
¶ Regioselectivity
Regioselectivity becomes a thing when we consider conjugate addition
- Simply by drawing out the resonance structure, we can see that there is two sites of attack
- Hard nucleophile tend to react to react at the carbonyl carbon of the enone (direct addition), while soft nucleophile tend to react with the beta carbon (conjugate addition)
- The most electron deficient carbon is the carbonyl carbon, so it favors an electrostatic attack (hard-hard interaction). The carbon has a large orbital, which makes it ideal for orbital interaction (soft-soft interaction)
If we want a direct addition, we can use a hard nucleophile
- Organolithium, Grignard and Organocerium are good choices
If we want a conjugate addition, we can use a soft nucleophile
- Organocopper and organocuperates are good choices
- A useful trick to get 1,4- selectivity with a Grignard reagent is to add a catalytic amount of Cu(I) salt
Sterics also play a role in regioselectivity
- If the steric hinderance is significant, a conjugate addition is more favorable
¶ Stereoselectivity
How to use the Felkin-Anh model
- Lable the size of the substituents adjacent to the carbonyl
- Choose a pair of anti-perriplanar as a reference point, this will be important later
- Draw the Newman projection and make sure the Large is perpendicular to the C=O bond
- The nucleophile attacks at the Bürgi-Dunitz angle, so we will want to choose the conformation where the nucleophile is approaching from the side of the small group
- We then obtain the Newman projection of the product
6.Using the pair of groups we chose in step 2, we shall rotate the C–C bond to make sure those two groups are anti-perriplanar again
- Convert the Newman projection back to the ordinary representation
¶ Chelate control may reverse selectivity
- When the substrate has an -heteroatom containing group with lone pairs available for coordination AND the reagent has a metal ion that can coordinate to the one heteroatom and the carbonyl, reaction can occur under chelation control
- This is because the negatively charged heteroatom and the carbonyl oxygen will coordinate to the metal ion together, which forces the oxygen to be unusually close to the large heteroatom
- Transition metals are common chelating cations, while main group metals are not
¶ Reduction
Hydride reagents as reducing agents
- Essentially transfer "" as a nucleophile, although be aware that a raw "" is NOT a nucleophilic (it is instead basic)
There are two subclasses of hydride reagents
- Nucleophilic hydride reagents acts as nucleophiles directly
- Electrophilic hydride reagents are electrophilic and require activation by a Lewis base before they can donate hydride
¶ Nucleophilic hydride reagents
Lithium aluminium hydride , LiAlH4
- VERY moisture sensitive, therefore used in DRY, aprotic solvents (e.g. Et2O)
- VERY reactive reducing agent and reduces most polarized functional groups (e.g. aldehydes, ketones, esters, nitriles, amides) relatively indiscriminately
- The mechanism:
- The coordination of the lithium ion to the carbonyl oxygen in the first step increases the reactivity significantly
- It is great for reducing amides to amine
- Here the iminium ion produced from collapse of the tetrahedral intermediate is much more reactive than the starting amide and so a second reduction produces the amine.
- It is great for partial reduction to aldehyde using the Weinreb amide
Sodiumborohydride , NaBH4
- Tolerates protic solvents, and MeOH or EtOH are usually used as the solvent
- NaBH4 is MUCH less reactive than LiAH4 and allow for the chemoselective reductio
- The mechanism:
- the protic solvent activates the carbony by hydrogen bonding. It therefore provides the proton for the alcohol product without the need for added water
Best for reductions of aldehydes and ketones to alcohols
¶ Electrophilic hydride reagents
Diisobutylaluminium hydride, DIBAL–H
- The mechanism:
- DIBAL–H first coordinates to the carbonyl oxygen to increase its own nucleophilicity (and also increase the electrophilicity of the carbonyl)
- Only until it is complexed by the carbonyl group ( Lewis base ) does it donates its hydride
- DIBAL–H is excellent for the partial reduction of esters
- Use of 1 equivalent. DIBAL-H at low temperature (usually at - 78oC, a temperature that is easily obtained using a dry ice/acetone mixture)
- The key to this is that at low temperature, the tetrahedral intermediate is stable, and so does not break down to liberate the aldehyde
Borane, BH3
- Chemoselectively reduce Carboxylic Acids over Esters
- The distinction arises from the rapid reaction of borane with the carboxylic acid to form an triacyloxyborane (which an ester cannot do due to absence of an acidic proton)
Lithium Borohydride, LiBH4~
- Chemoselectively reduce Esters over Carboxylic Acids
-LiBH4 – as an evidently more powerful reducing agent than NaBH4 as it DOES reduce esters, but does not react with carboxylic acids
Dissolving metal reductions
- Dissolve zero valent metals (e.g. Na) in ammonia to give solvated electrons
- Mechanistically, it involves the step-wise addition of electrons and protons, where added alcohol provides the protons
Reductive amination of carbonyls to amines
- The reaction of an aldehyde or ketone with a primary amine under acidic conditions in the presence of Na(CN)BH3 gives a secondary amine
- Na(CN)BH3 is basically a deactivated NaBH4 due to the electron withdrawing cyano group. This is important because it only reacts with the most electrophilic species ( iminium ion ) and does not reduce the less reactive carbonyl or imine
Reduction of ketones to alkanes
- Random derivation-less, proof-less,arbitrary fact
Reduction of nitriles
- We can reduce nitriles to primary amines using LiAlH4 or DIBAl-H at room temperature or catalytic hydrogenation
- The use of DIBAL-H at low temperature can again allow partial reduction, leading to the aldehyde after hydrolysis.