¶ Conformations
¶ Conformation of Carbon Chains
The shape of molecules are not static due to the free rotation about the sigma bond
- Sigma bonds are cylindrically symmetric, which allows for free rotation
- The extra energy present in certain conformations are called torsional strain
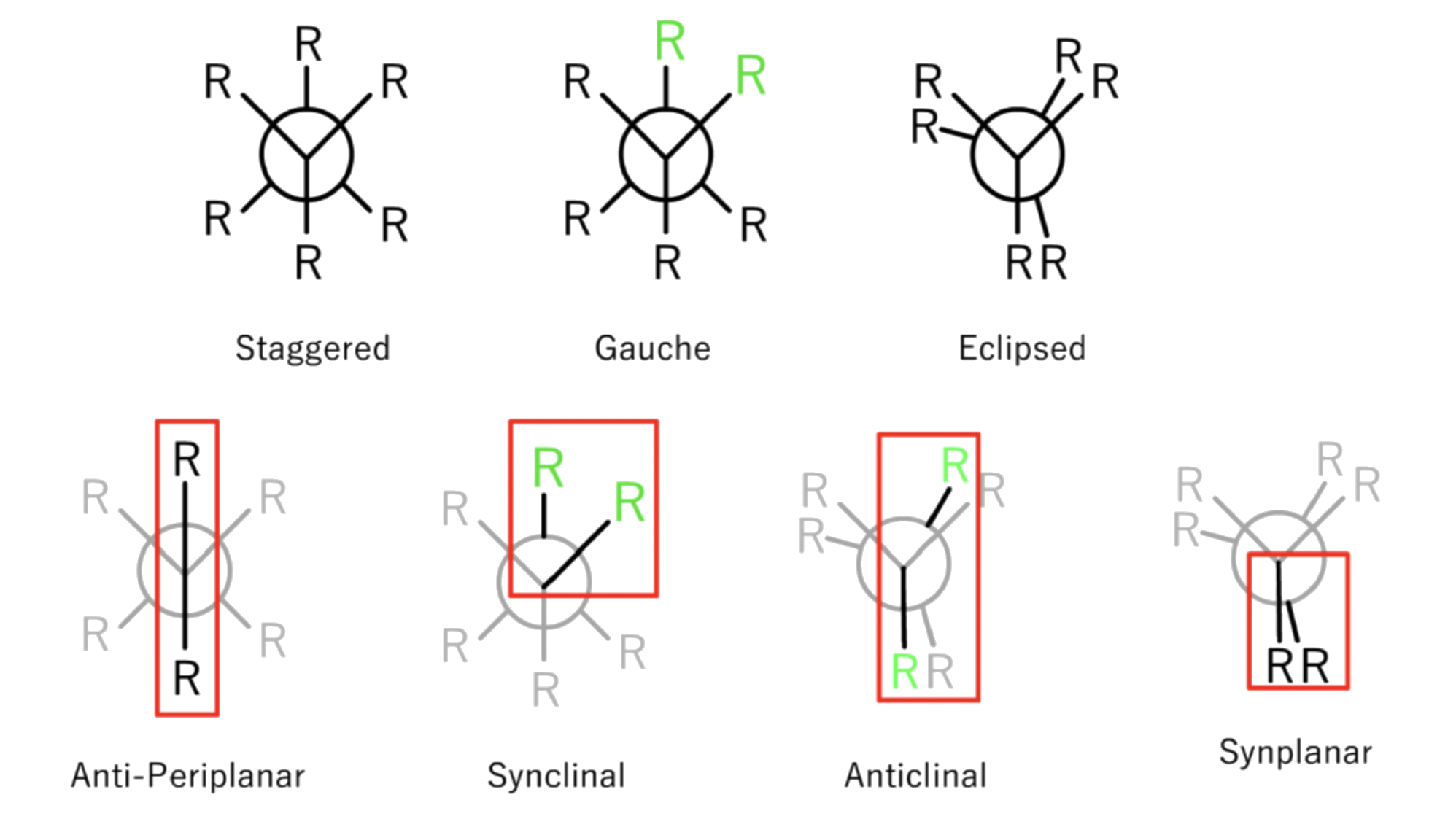
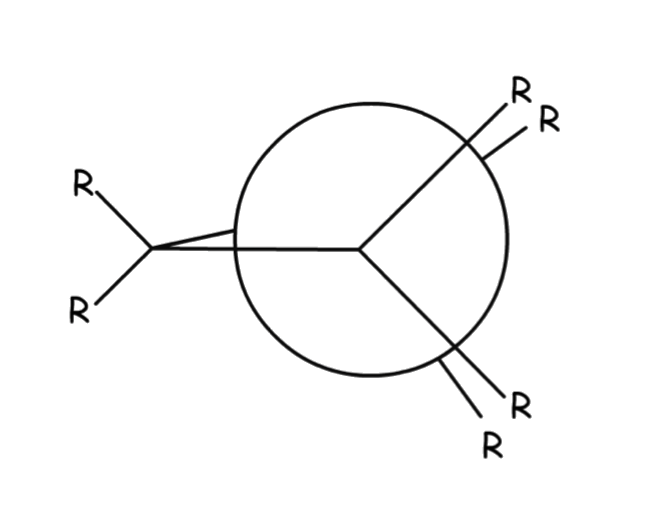
The conformations have different names depending on the dihedral angle
- Dihedral angle is the angle between C-X bonds on front and back carbons as viewed end-on
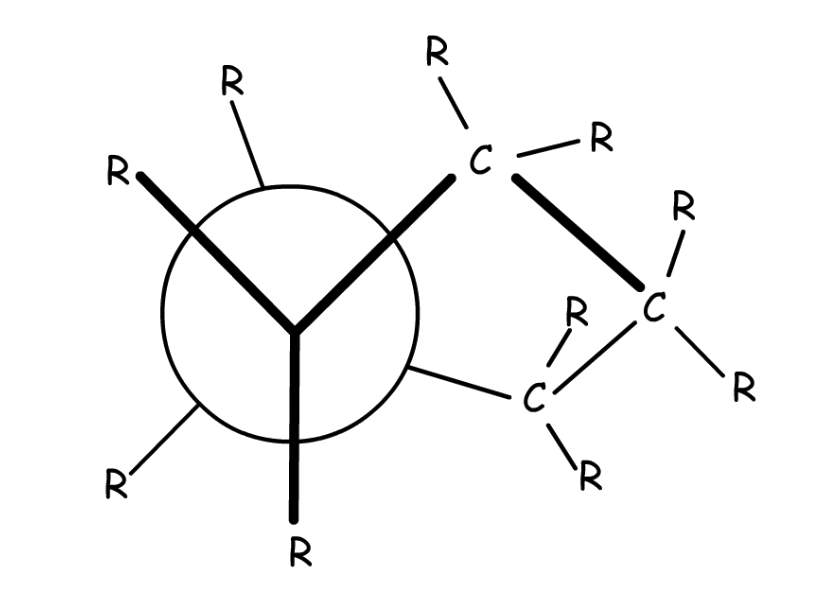
The staggered conformation has a lower torsional strain than the eclipsed one for several reasons:
- The eclipsed conformation has a high steric hinderance
- In the eclipsed conformation, there is a steric clash between the R groups, which destabilizes the conformation. (minor factor if the R group is small)
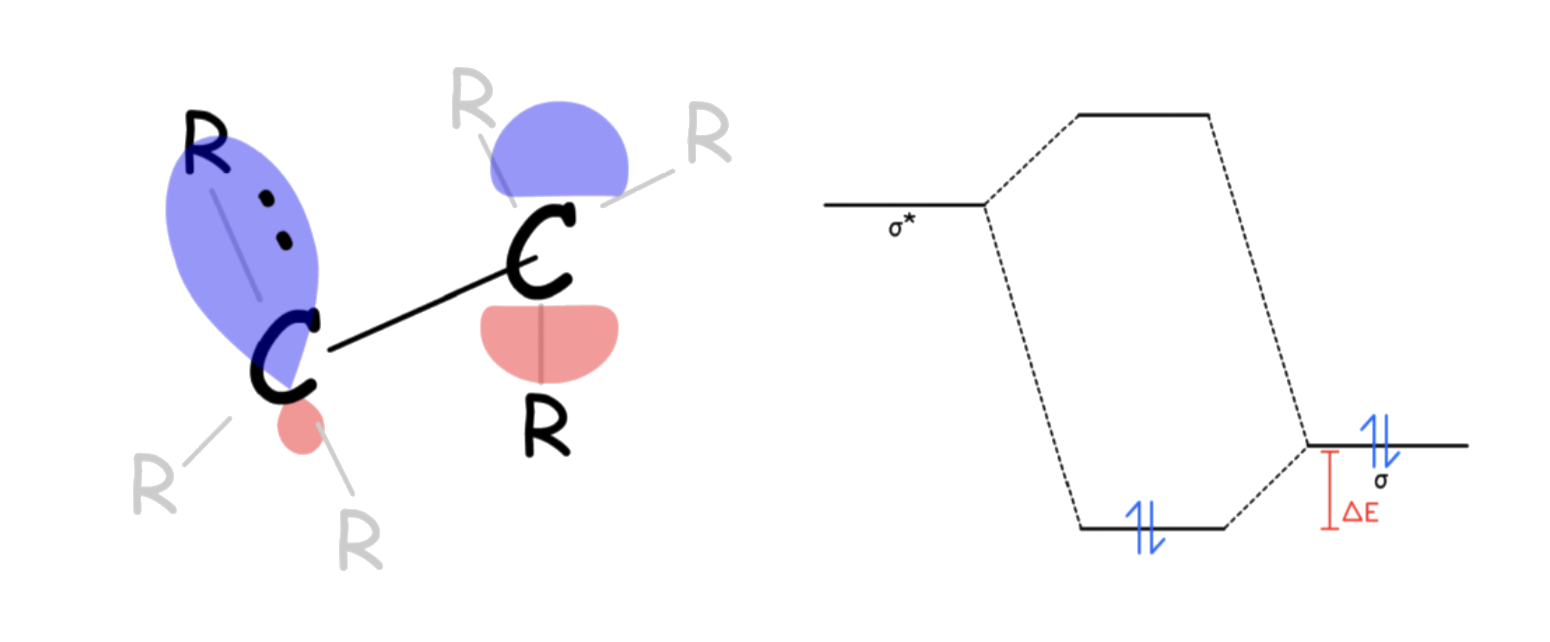
- In the staggered conformation, the molecule can be stabilized through negative hyperconjugation
- The sigma bonding orbital of the C-R1 bond and the sigma antibonding bonding orbital are of the same symmetry when aligned correctly
- Negative Hyperconjugation can only occur when the two groups are at ( anti-periplanar) to each other
- Negative hyperconjugations most commonly observed when the σ* orbital is located on certain C–F or C–O bonds, and does not occur to an appreciable extent with normal C–H bonds
- Electronegative atoms lower the energy of MOs, including the antibonding orbitals. Since orbitals closer in energy mix better, by lowering the energy of the antibonding orbital, the effect of negative hyperconjugation is more prominent
¶ Conformation of Carbon Rings
The strain energy is the extra energy ( relative to a strain-free molecule ) in the molecule due to strain
- Strain energy is usually a combination of torsional, angle and transannular strain
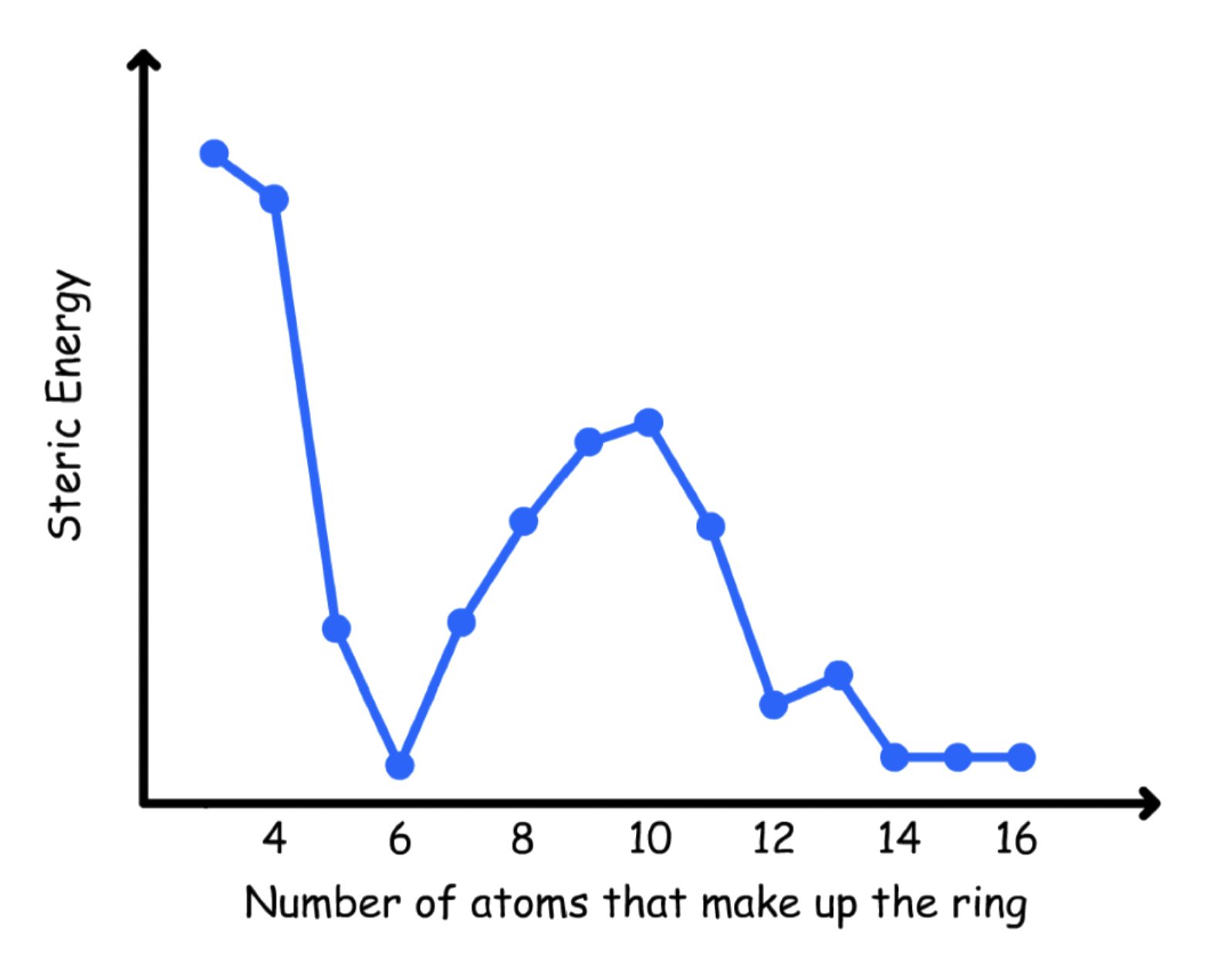
Larger rings are flexible enough to avoid strains
- 6-membered rings and rings of 14 carbons or more are strain-free
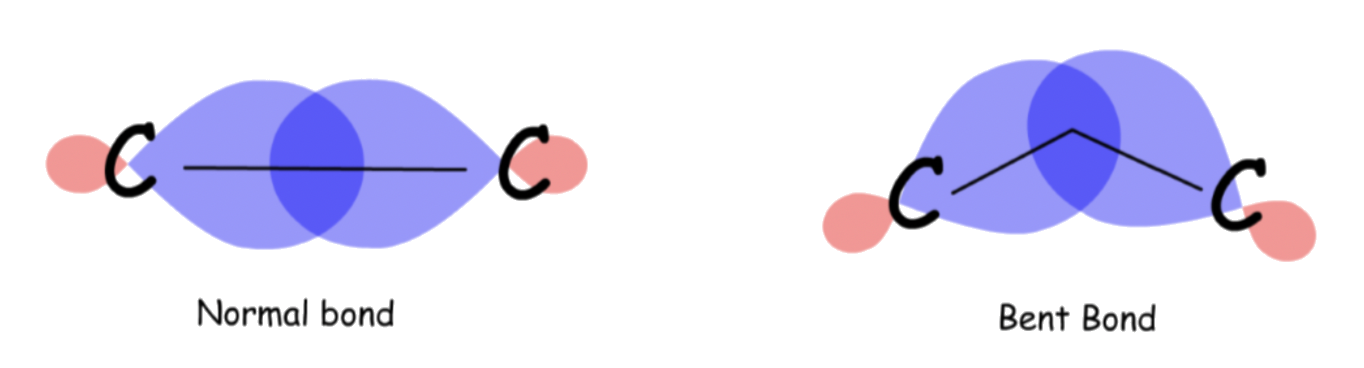
3-membered rings
3-membered rings are rigid, planar molecules because three points ( The ring atoms ) define a plane
- No rotation around the C–C bonds
- Only one conformation for each molecule
3-membered rings are the most strained of all rings
- The angle strain caused by its 60° C–C–C bond angles
- There's significant torsional strain because all the neighboring carbons are in the eclipsed conformation
To account for such a drastic deviation from the ideal bond angle ( 109.5° ), the C-C bonds in the ring is bent
- In an unstrained alkane, maximum bonding is achieved when two atoms have their overlapping orbitals pointing directly to each other. However, in 3-membered rings, the orbitals overlap at a slight angle
- The result is that cyclopropane bonds are weaker and more reactive than typical alkane bonds
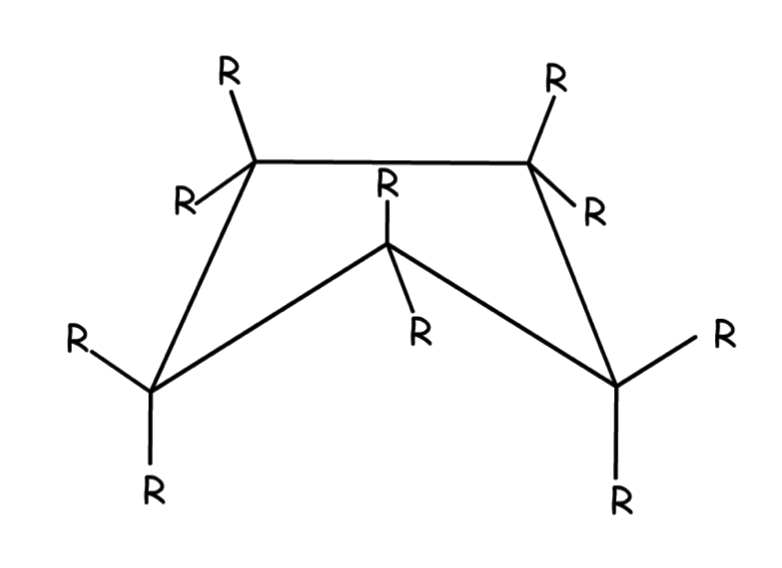
4-membered rings
4-membered rings has less angle strain than cyclopropane, but has more torsional strain
- There's a larger number of ring substituents/ hydrogens, which results in a greater degree of torsional strain
4-membered rings are not flat, but is slightly bent
One carbon atom lies about 26° above the plane of the other three
- The slight bent increases the angle strain ( flat: 90° to slightly bent: 88.5° ) but decreases the torsional strain, until a minimum-energy balance between the two opposing effects is achieved
5-membered rings
Although a flat 5-membered ring has practically no angle strain, it has a large amount of torsional strain
- 5-membered rings twist to adopt a puckered, non-planar conformation that strikes a balance between increased angle strain and decreased torsional strain
5-membered rings have two stable puckered conformations
- Envelope
- One of the carbon is flipped up, while the other four carbons are approximately on the same plane
- MOST of the substituents/ hydrogens are NEARLY staggered with respect to their neighbors
- Half-Chair
- Two opposite carbons flip up while the remaining three carbons form a triangular plane
- MOST of the substituents/ hydrogens are NEARLY staggered with respect to their neighbors
The two puckered conformations are similar in stability in the absence of substituents. When the ring has a single substituent, the envelope conformation becomes slightly more favorable
6-membered rings
Six-membered rings have no strain energy
- There are no angle strain and torsional strain
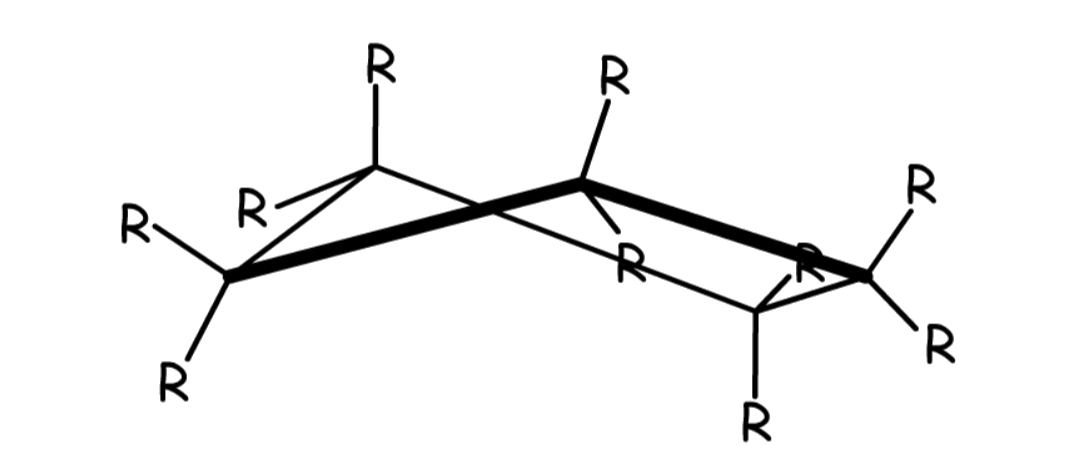
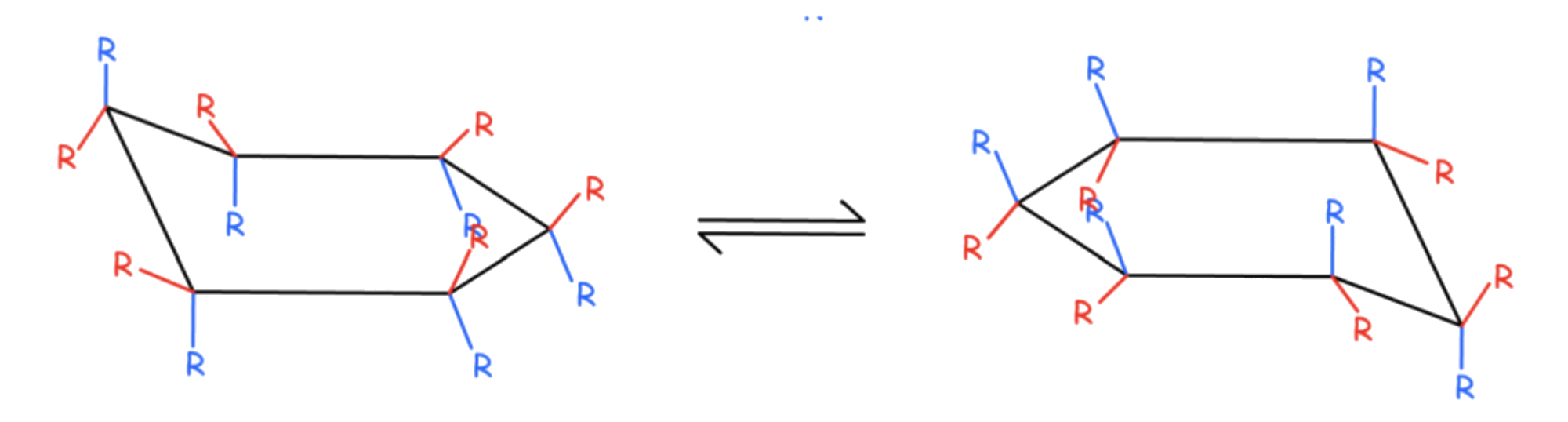
6-membered rings have four stable conformations
- Chair
- Every carbon in the chair conformation is staggered with respect to its neighbor
- There is no strain energy
- The most stable conformation
- Twist-boat
- It is also nearly free of angle strain
- It has both steric and torsional strain higher the chair conformation
- Second-most stable conformation
- Boat
- The boat conformations have higher energy than the twist-boat conformations
- The interaction between the two flagpole hydrogens, in particular, generates steric strain
- Torsional strain exists between the C2–C3 and C5–C6 bonds, they are staggered with respect to each other (carbon number 1 is one of the two on a mirror plane)
- The boat conformations spontaneously distorts to twist-boat conformations
- Half-Chair
- One carbon is flipped up, while the other five carbons form a relatively flat plane
- Partial plane conformation
- In the planar portion of half chair, the C-C bond angles are forced to 120° which creates significant amounts of angle strain
- The carbons in the planar portion of the half chair are fully eclipsed, which amounts to a high torsional strain
- The out-of-plane carbon allows for some of the ring's bond angles to reach 109.5° and for some of C-R bonds to not be fully eclipsed
- The most unstable conformation
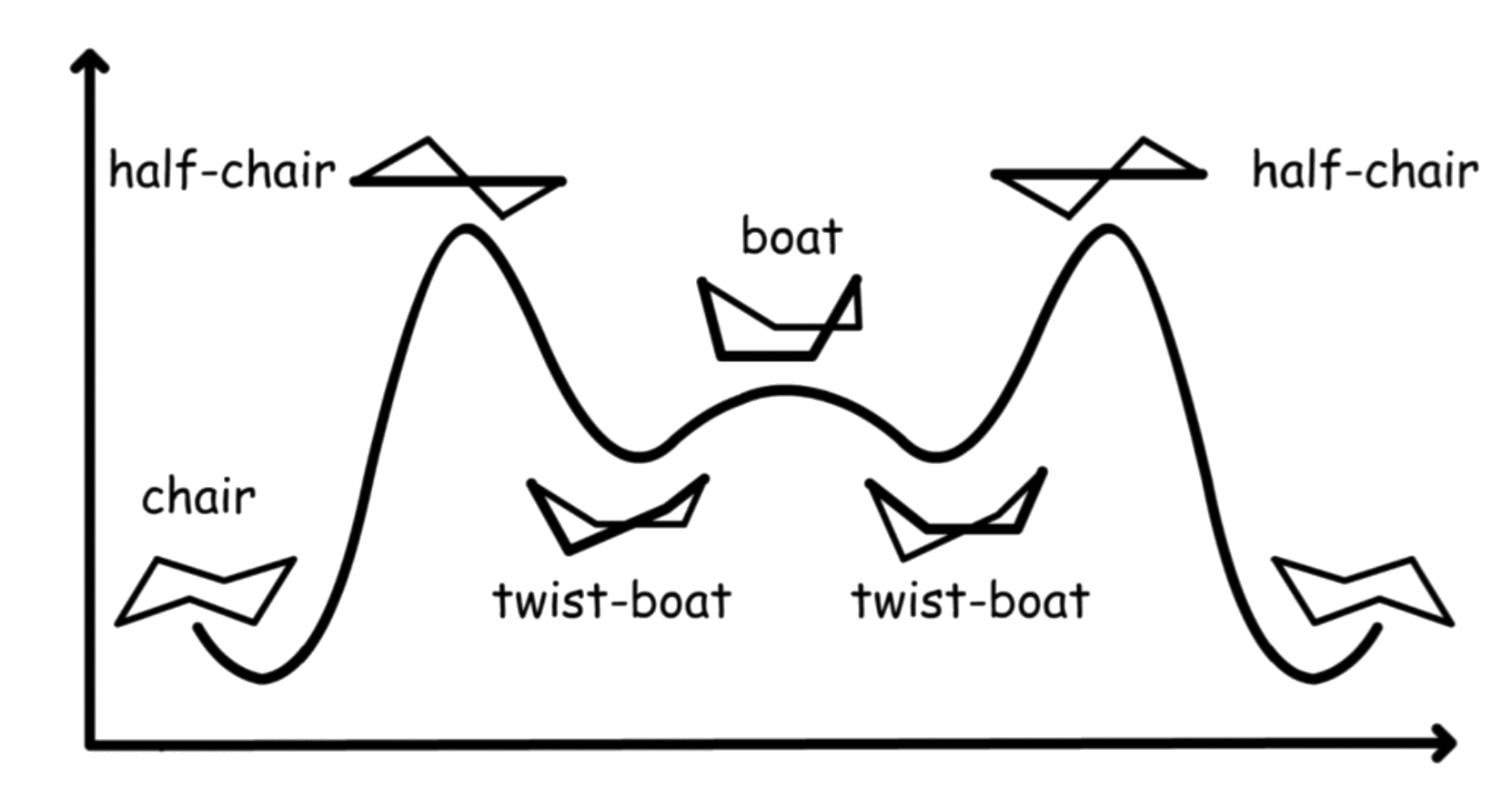
The reaction coordinate of all the conformations:
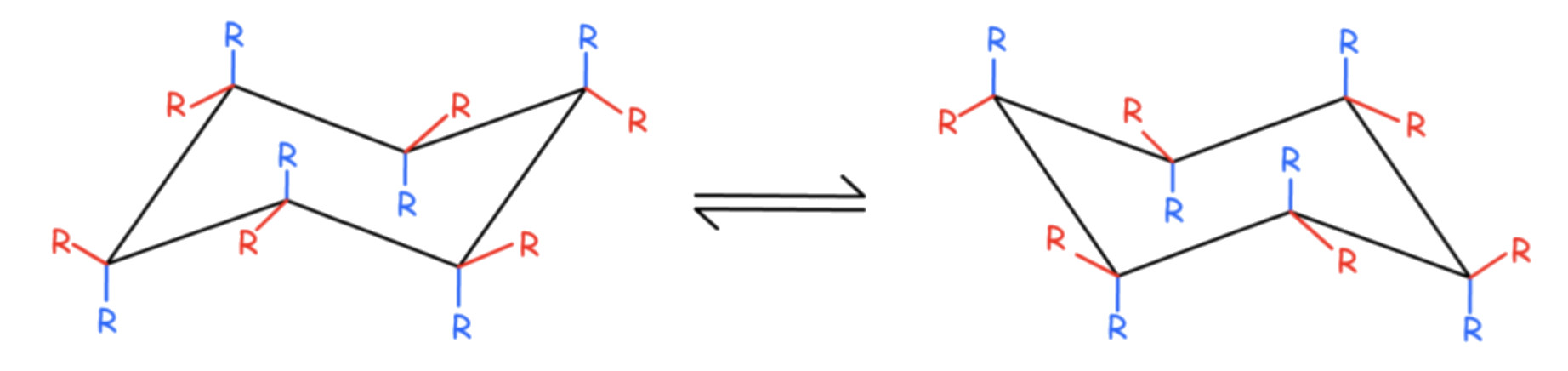
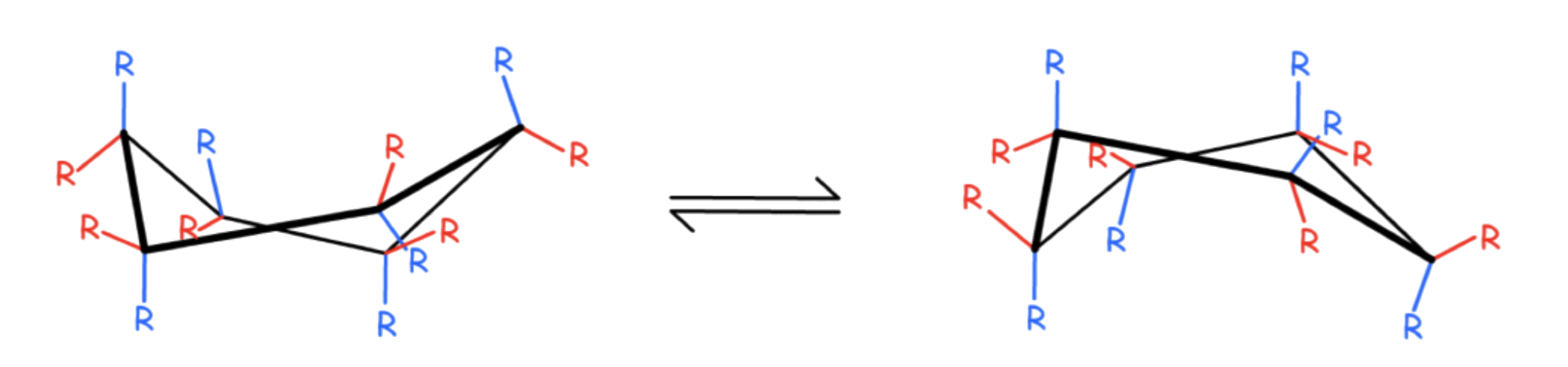
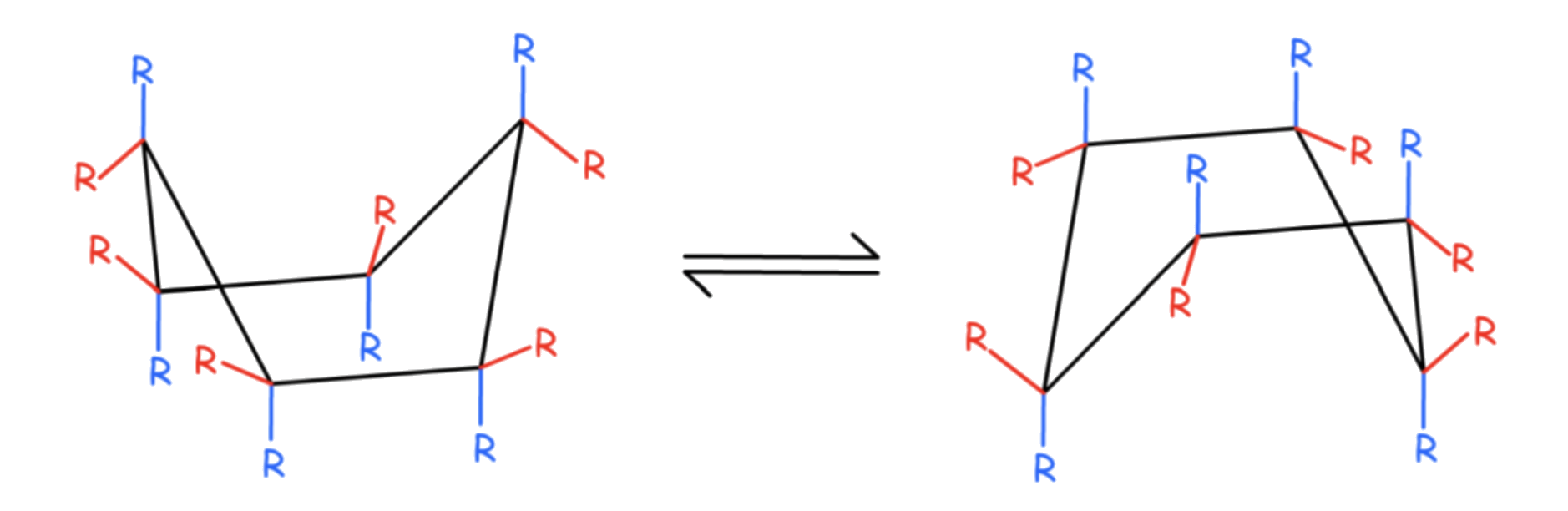
The interconversion between the two chair conformations involves the following sequence: chair → half-chair → twist-boat → half-chair′ → chair′
The boat conformation is a transition state, allowing the interconversion between two different twist-boat conformations
- The boat conformation is not necessary for interconversion between the two chair conformations of cyclohexane
- It is often included in the reaction coordinate diagram used to describe this interconversion because its energy is considerably lower than that of the half-chair, so any molecule with enough energy to go from twist-boat to chair also has enough energy to go from twist-boat to boat
there are multiple pathways by which a molecule of cyclohexane in the twist-boat conformation can achieve the chair conformation again.
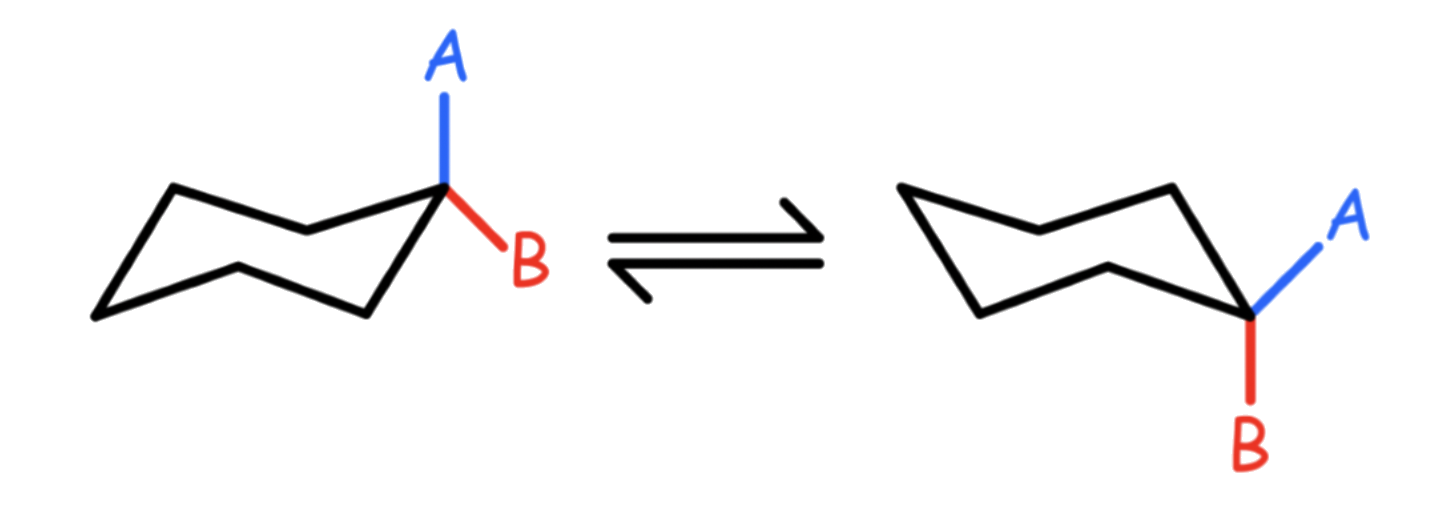
There are two types of substituents/ hydrogens in the molecule: axial and equatorial
- For large substituents, the equatorial position is more stable as it points away from the other hydrogens/ substituents.
- The molecule can undergo 'ring flipping' to change such that the larger substituent is more likely to be in the equatorial position
¶ Unsaturated Carbons
¶ Alkene
An alkene, sometimes called an olefin, is a molecule that contains carbon-carbon double bond(s)
- Those carbons are sp2 hybridized, so they both adopt a trigonal planar geometry
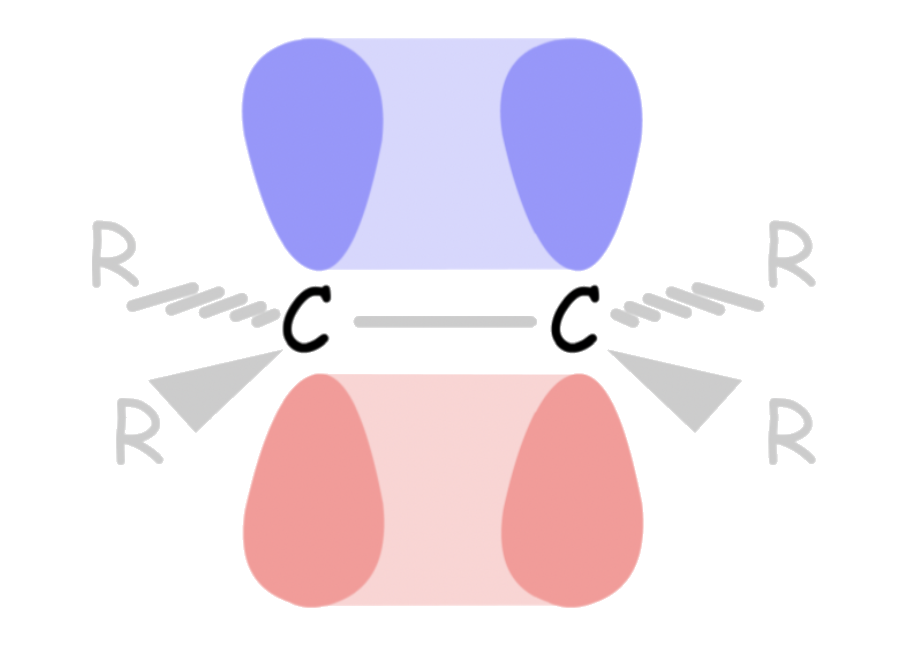
- The double bond of alkene is formed by the side-to-side overlap of two p-orbitals
Stereochemistry of alkenes
Double bonds cannot be rotated
- Rotation about the double bond requires the breaking of π bond
- The lack of free rotation about the carbon-carbon double bond gives rise to diastereomers
Cis-trans isomerism is not limited to disubstituted alkenes, it can occur whenever both double-bond carbons are attached to different groups
- If one of the double-bond carbons is attached to two identical groups, cis-trans isomerism is not possible
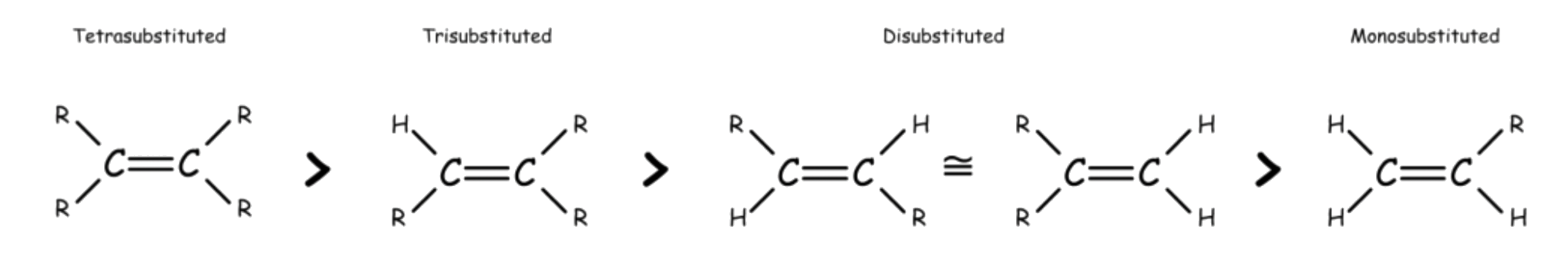
Stability of Alkenes
Cis-alkenes are less stable than trans-alkene due to steric hinderance
The stability of alkene increases with the order of substitution
The stability order of substituted alkene is due to a combination of two factors
- Bond Strength
- A σ bond between an sp2 carbon and an sp3 carbon is somewhat stronger than a σ bond between two sp3 carbons
- The number of sp2–sp3 σ bonds increases with the degree of substitution
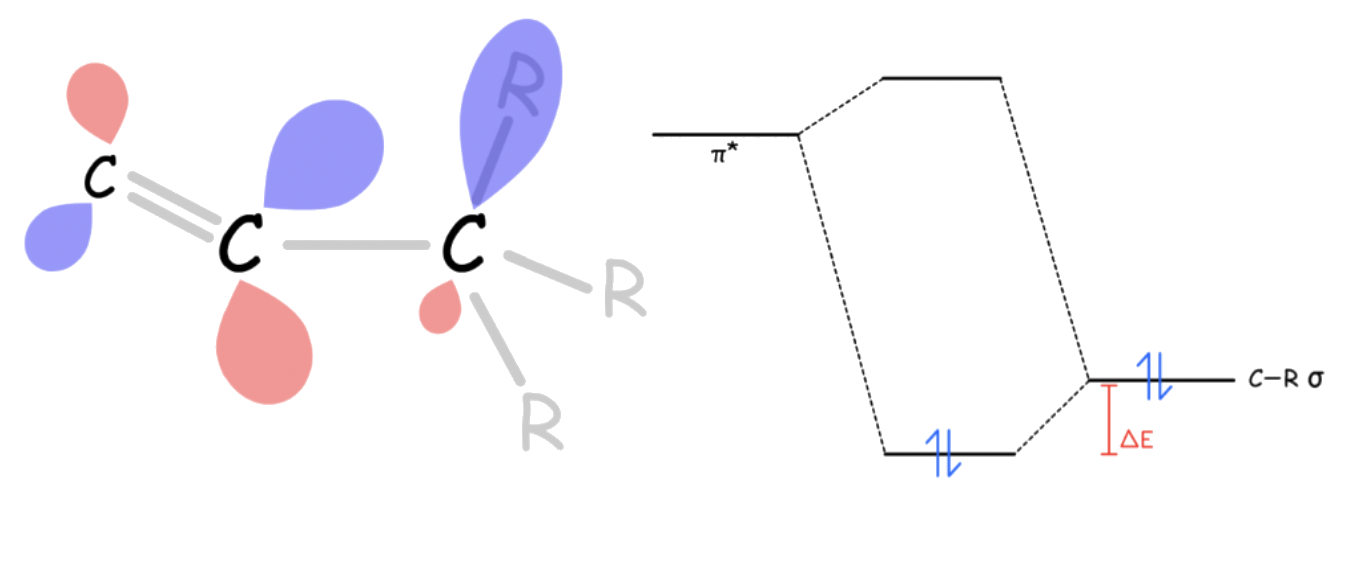
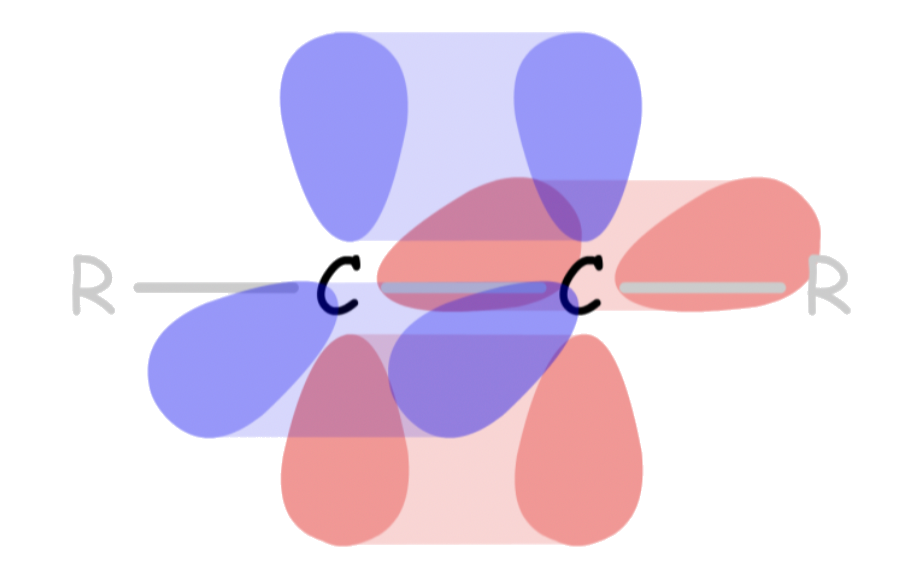
- Sigma-conjugation
- A stabilizing interaction between a π* orbital and a properly oriented σ orbital on neighboring carbons that are roughly parallel to the p orbital
¶ Alkyne
The functional group of an alkyne is a carbon-carbon triple bond
- Those carbons are sp3 hybridized, so they both adopt a linear geometry
The triple bond of alkene is formed by two sets of side-to-side overlap of two p-orbitals orthogonal to each other
Alkyne are similar to alkene in that the multiple bond in each is a combination of sigma and pi bonds
- Both type of functional groups undergo similar chemical reactions
Acidity of Terminal Alkynes
An important reaction of alkynes that is not typical of alkene is the conversion of terminal alkynes to their alkali metal salt
- These salt are good nucleophiles
- They are valuable building blocks for the formation of carbon-carbon bonds through nucleophilic substitutions
Terminal alkynes, alkynes with carbon(s) bonded to a hydrogen, are acidic
- A hydrogen bonded to a triply bonded carbon atom of a terminal alkyne is sufficiently acidic that it can be removed by a strong base

¶ Oxygenated Functional Group
¶ Carbonyl Compounds
Carbonyl compounds are molecules that contain carbon-oxygen double bond(s)
- The carbon and oxygen are sp2 hybridized, so they both adopt a trigonal planar geometry
- The double bond of the carbonyl functional group is formed by the side-to-side overlap of two p-orbitals
Carbonyl compounds refer to the two families of compounds known as aldehydes and ketones, both of which contain the carbonyl functional group:
- For aldehydes, at least one of the R group is a hydrogen atom
- For ketones, both R groups must be alkyl or aryl groups that may or may not be the same
The carbonyl group has characteristic properties that are exhibited by both classes of compounds such that the two homologous series are more conveniently considered together
- However, the attachment of a hydrogen to the carbonyl group of an aldehyde does causes the two families to be different in certain ways
Electrophilicity of Aldehyde and Ketone
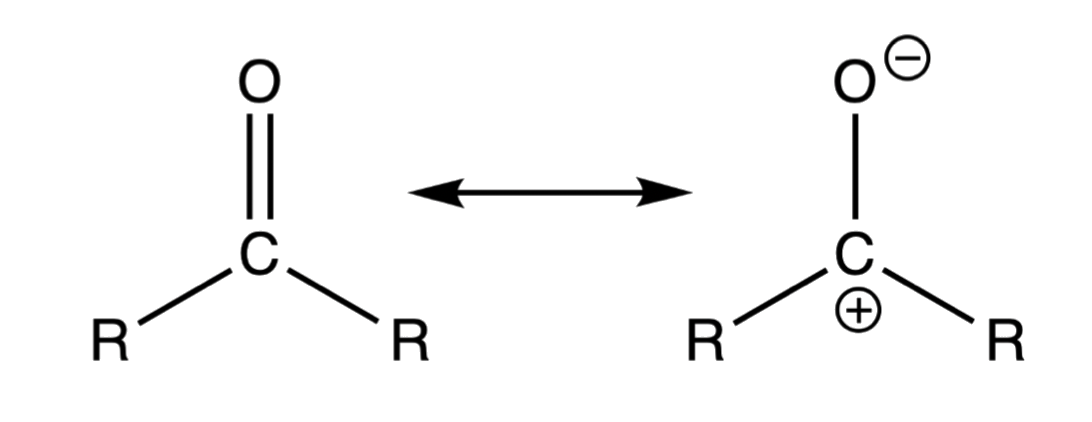
Within the carbonyl functional group, the electrons in the σ and π bonds are drawn towards the more electronegative oxygen atom
- The carbonyl oxygen bears a substantial partial negative charge, whereas the carbonyl carbon bears a substantial partial positive charge
- In addition, the resonance structure emphasizes the reactivity of the carbonyl carbon as a electrophile and the carbonyl carbon as a nucleophile
- Despite having the same electrophilic site ( carbon ), their electrophilicity differs quite a bit
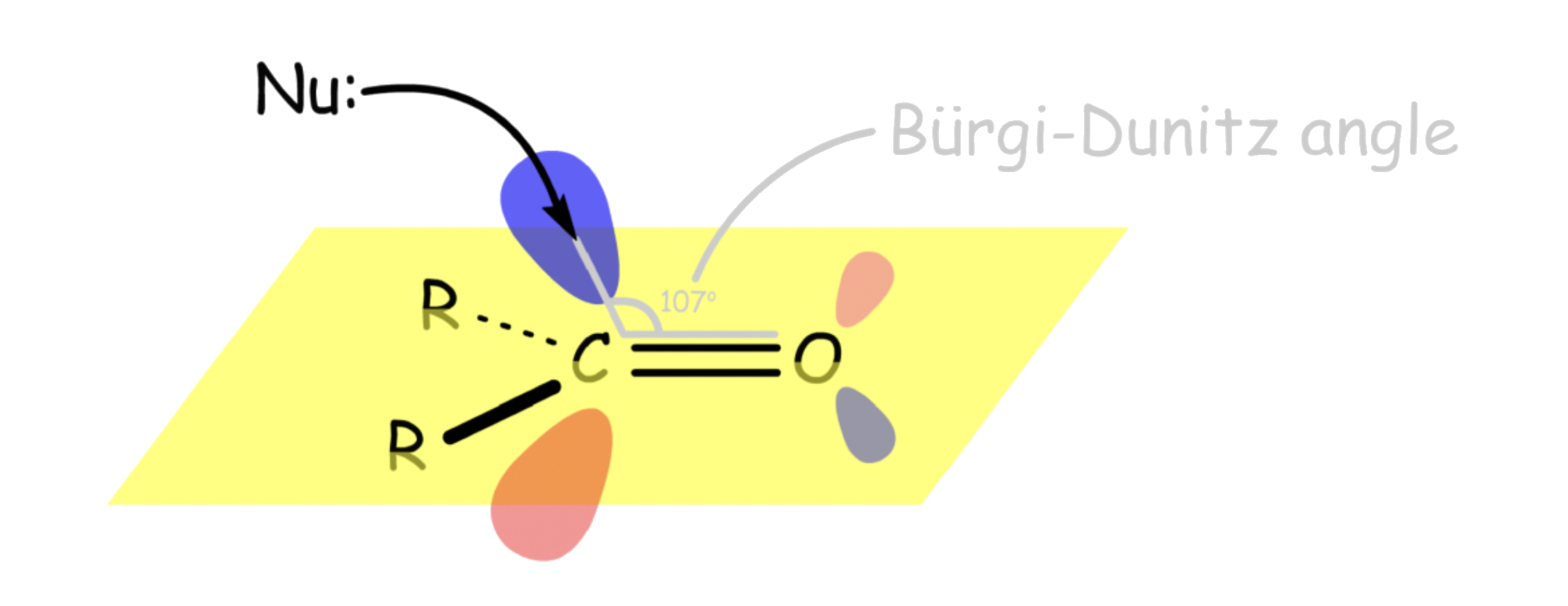
For both aldehydes and ketones, the nucleophile will attack the carbon through the π* orbital
- The nucleophile attacks at about 107° relative to the plane ( ∠O C Nu ) of the molecule. This angle is called the Bürgi-Dunitz angle
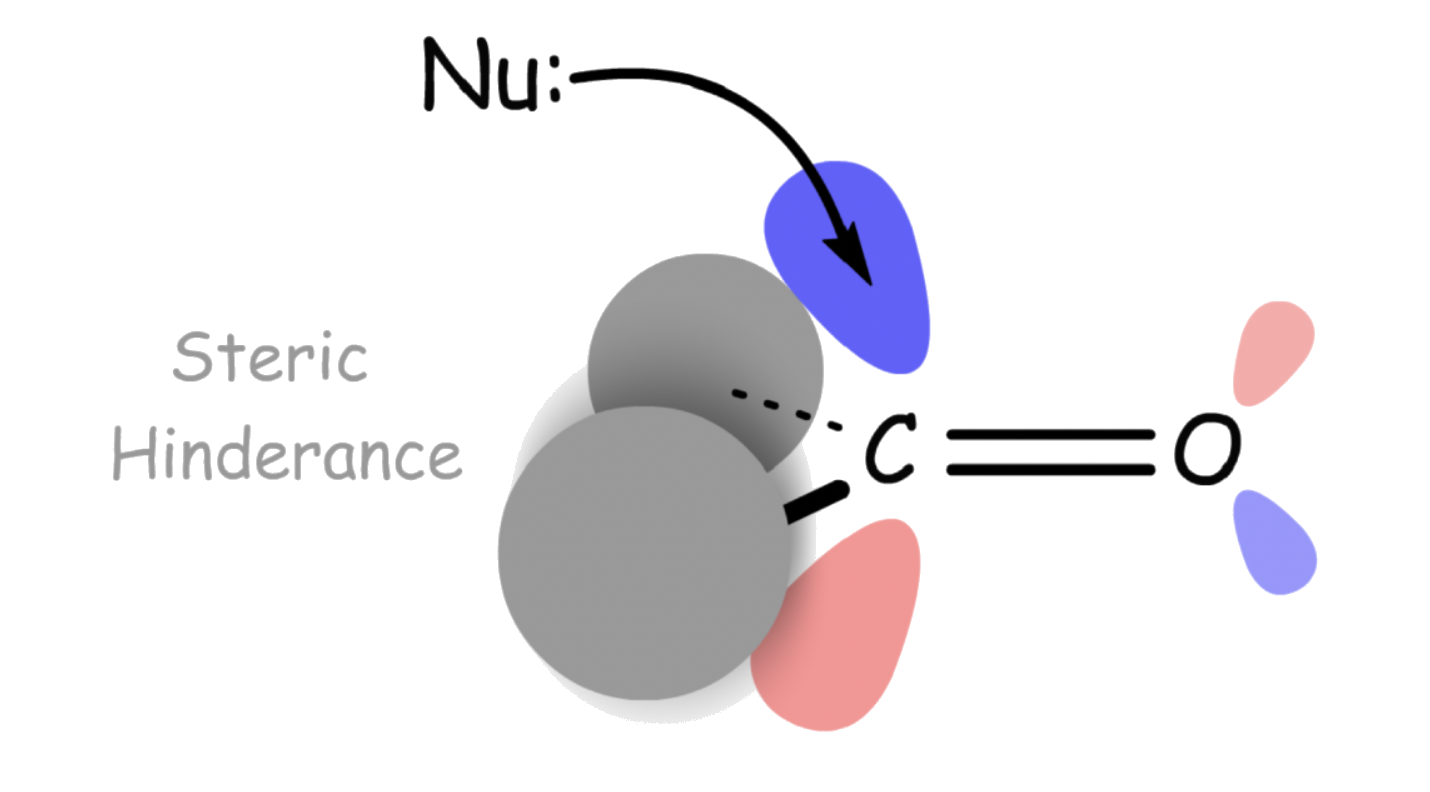
In general, aldehydes are more electrophilic than ketones. This is a combination of electronic and steric factors
- Steric Reasons:
- For aldehydes, the hydrogen is relatively small, so it will not block the entrance of the nucleophile
- For ketones, there are two R groups that prevents the approach of the nucleophile. This becomes an even bigger problem for bulky nucleophile
- Electronic Reasons:
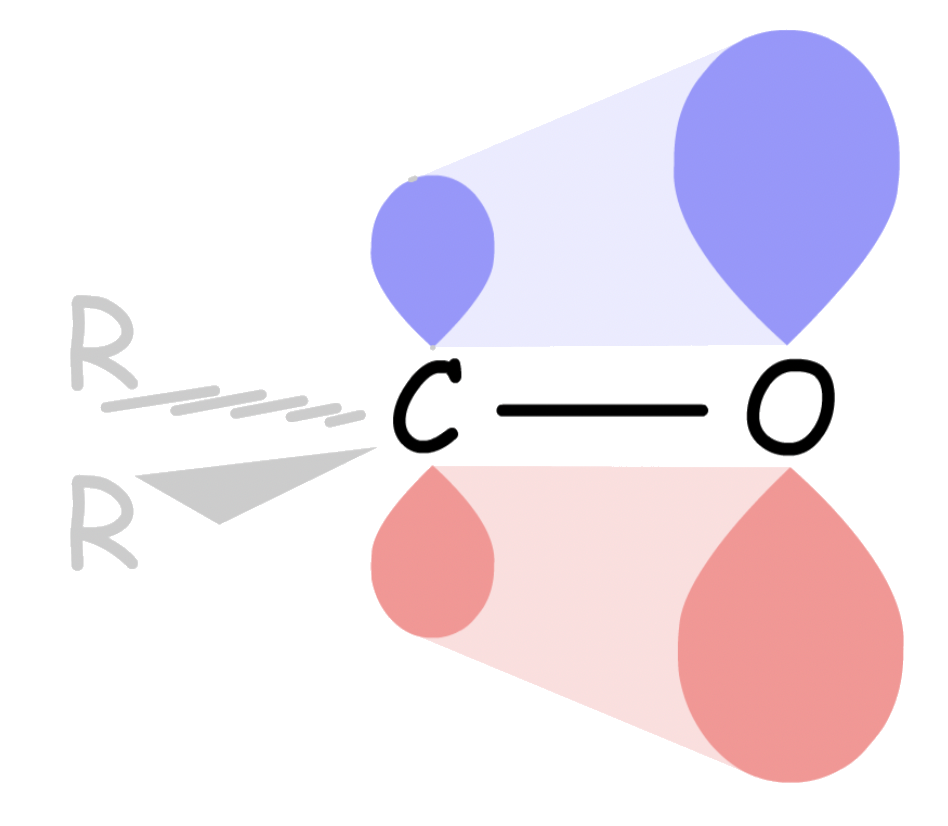
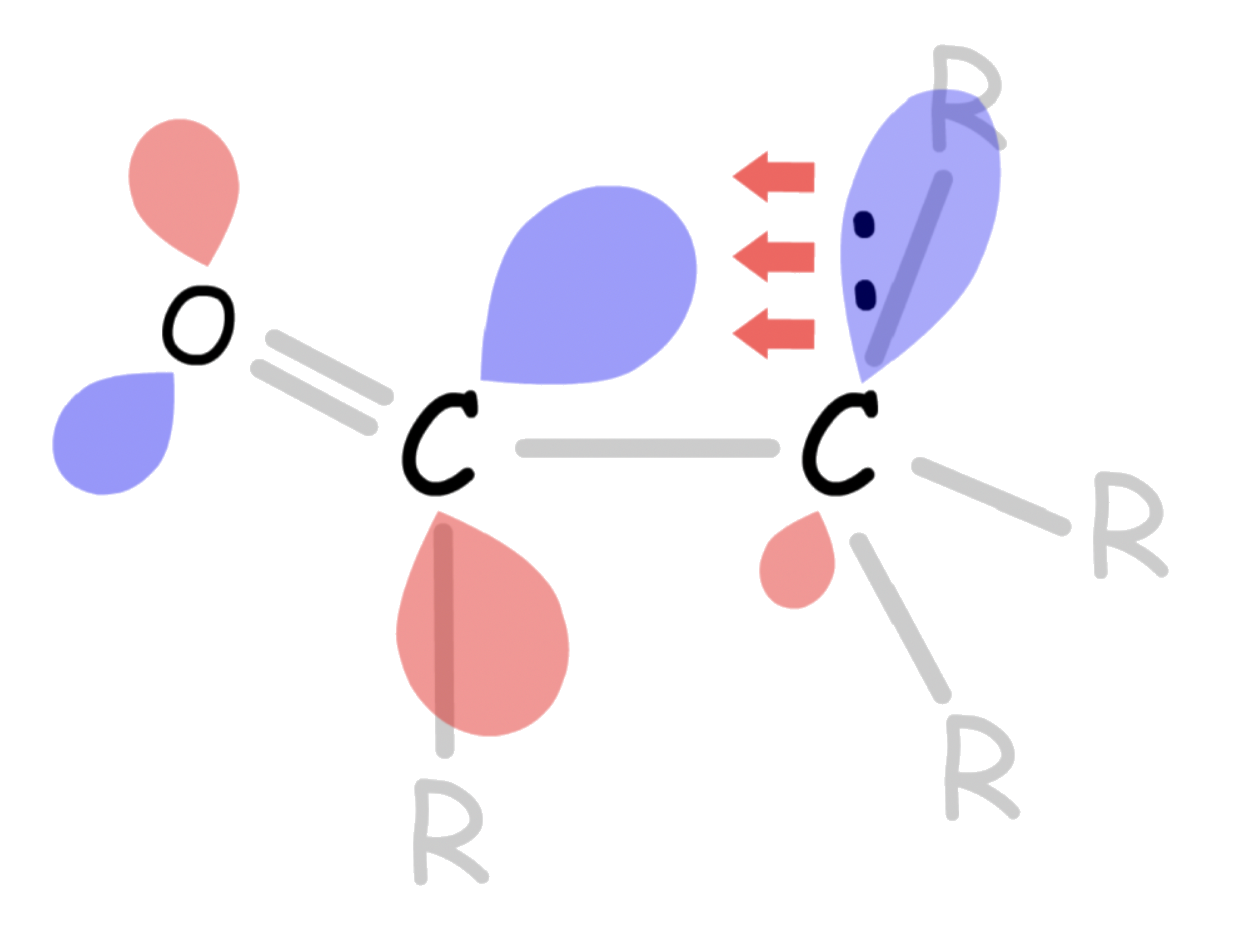
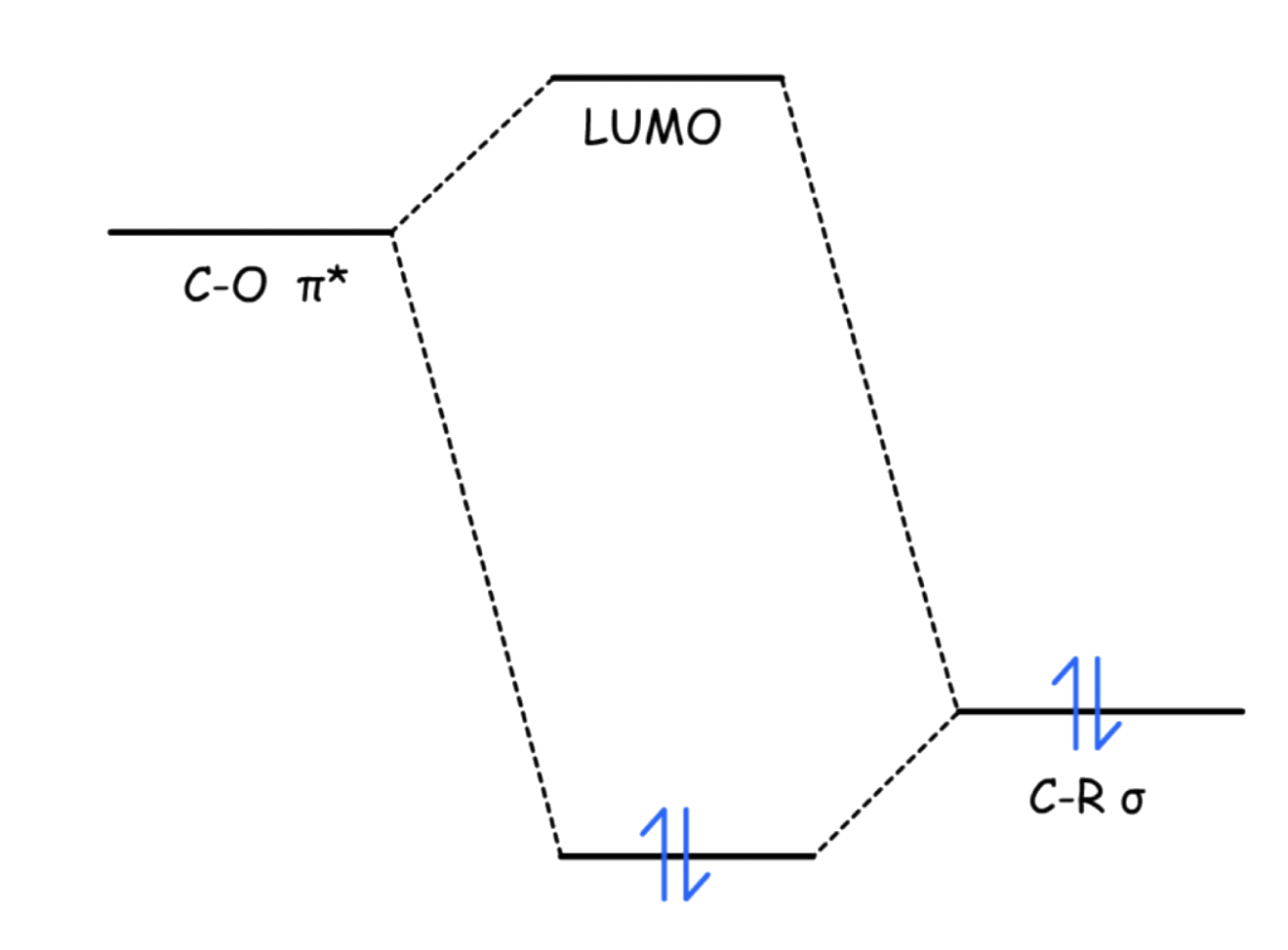
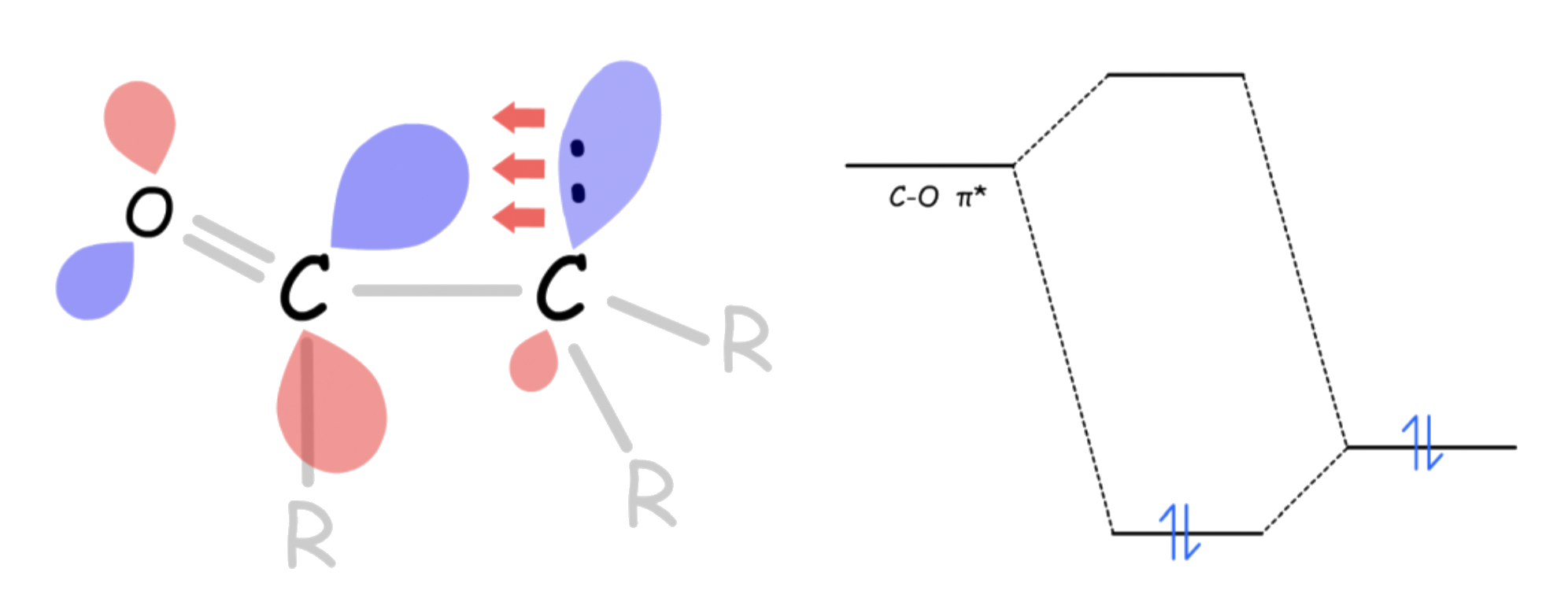
- One way to interpret this is that the C–R σ orbital is donating electron density towards the empty C–O π* orbital. This makes the C–O π* orbital less electron-deficient, thus less electrophilic
- A more accurate depiction would be that the adjacent C–R σ orbital mixes with the C–O π* orbital, causing the LUMO to be higher, which is less accessible to the nucleophile
¶
Acidity of α-hydrogen
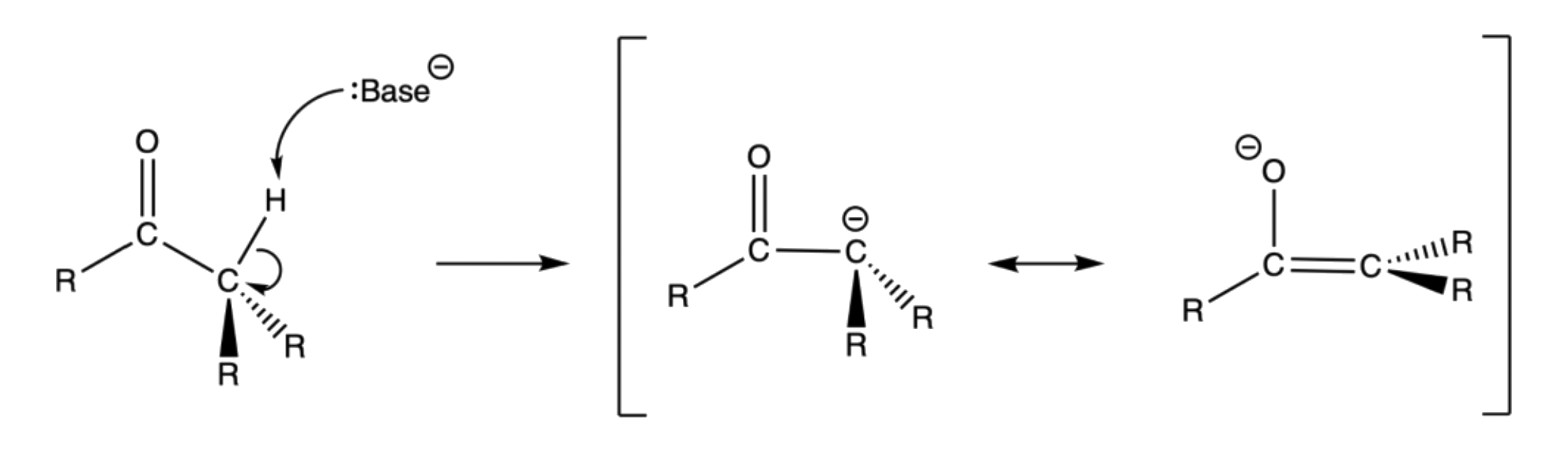
A carbon atom adjacent to a carbonyl group is called an α-carbon, and the hydrogen(s) bonded to it are called α-hydrogens
- Alkyl hydrogen atoms bonded to a carbon atom in a α position relative to a carbonyl group display unusual acidity
α hydrogens are weakly acidic because the conjugate base, called an enolate, is stabilized though conjugation with the π orbitals of the carbonyl
- From a molecular orbital perspective, the orbital mixing between the filled sp3 orbital and the empty C–O π* orbital of the conjugate base allows the electron pair to be pulled deeper in energy level, thus more stable
- It is worth noting that if the nucleophilic attack is hindered enough, the carbonyl compounds may take part in an acid-base reaction instead ( This usually happens to ketones )
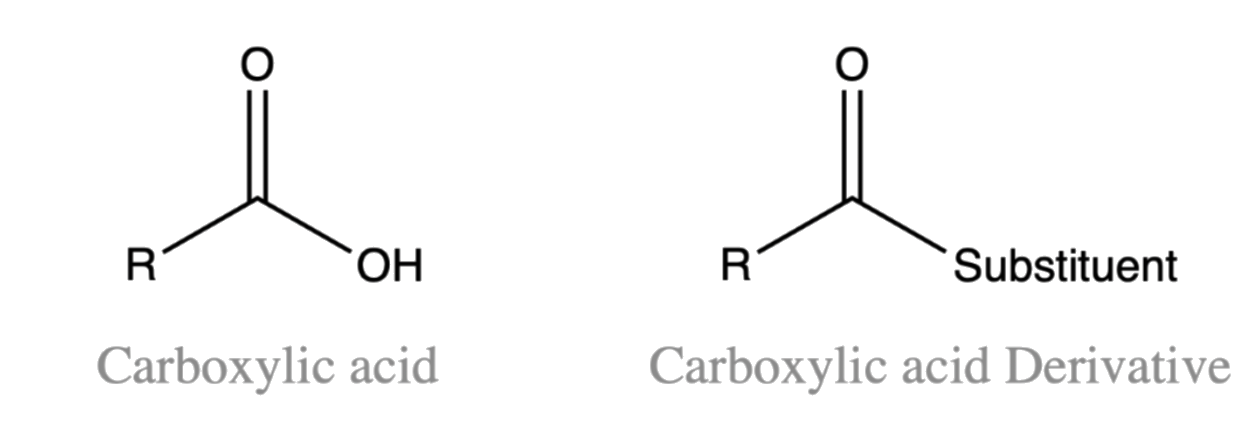
¶ Carboxylic Acid and Its Derivatives
The important classes of organic compounds known as alcohols, phenols, ethers, amines and halides consist of alkyl and/or aryl groups bonded to hydroxyl, alkoxyl, amino and halo substituents respectively
- If these same functional groups are attached to an acyl group ( R–C=O) their properties are substantially changed
- These compounds are designated as carboxylic acid derivatives
Carboxylic acids have a hydroxyl group bonded to an acyl group
- By the replacement of the hydroxyl group with substituents, such as halo, alkoxyl, amino and acyloxy, we will get the carboxylic acid derivatives
The substituent that replaces the OH must not be an alkyl group or a hydrogen atom
- Alkyl groups and hydrogen atom lack the ability to undergo sophisticated orbital mixing with the acyl group to give the common properties of carboxylic acid derivatives
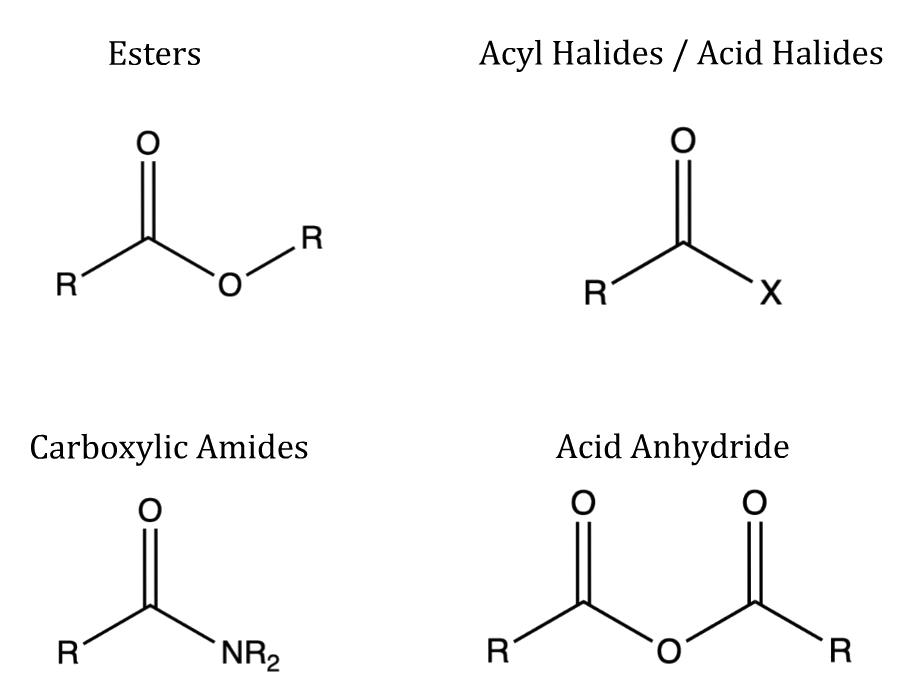
There are four main groups of carboxylic acid derivatives
¶
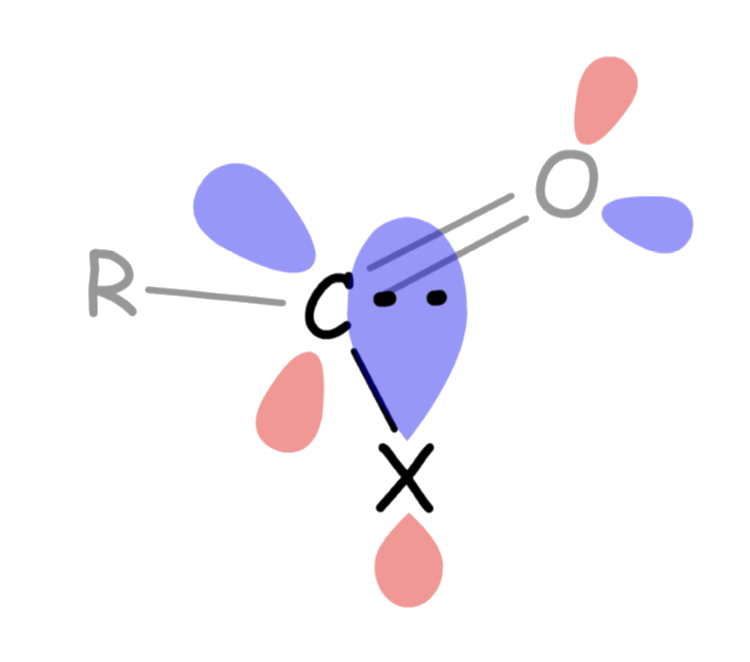
Orbitals Mixing of the acyl group and substituent
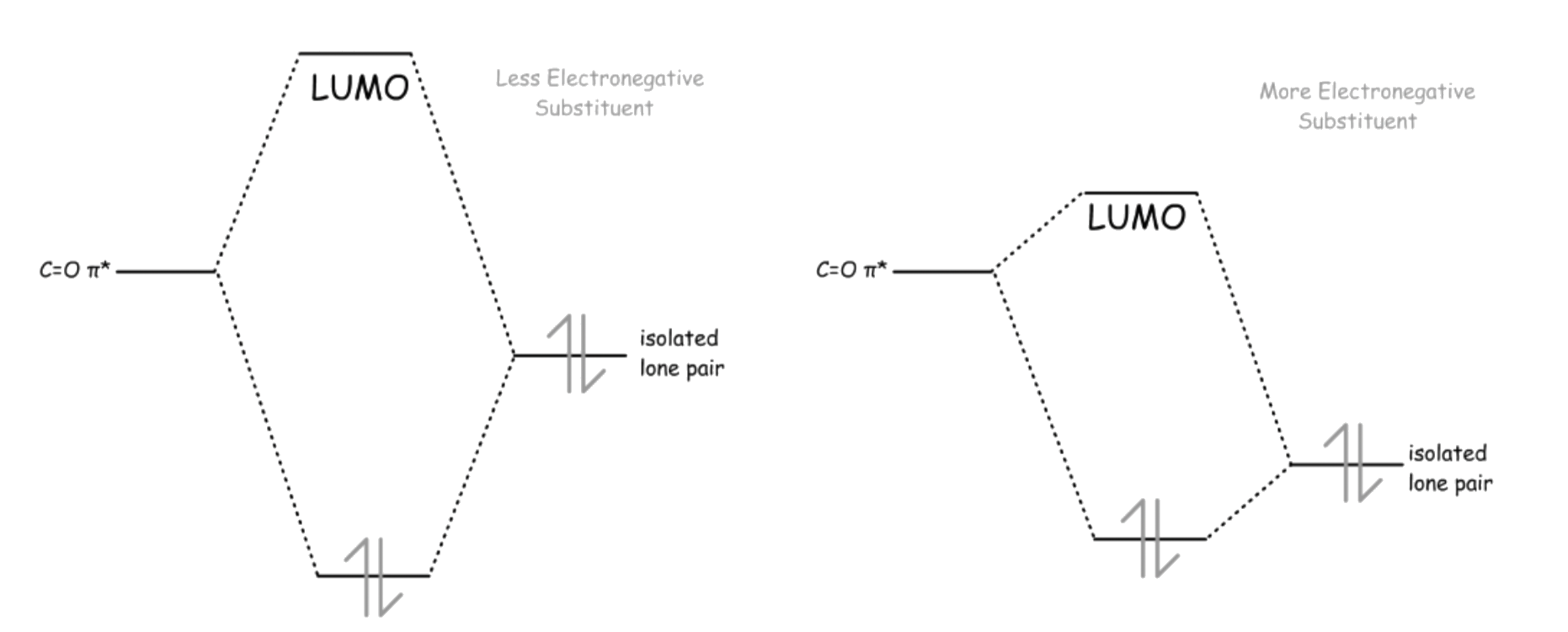
All substituents that are attached to the carbonyl carbon possesses at least one lone pair electrons, which allows it to delocalize between the substituent and the acyl group
- The lone pair of the substituent is delocalized with the π* orbital of C=O
- Since electron density is donated to the antibonding orbital of CO, the bond order of CO decreases due to this effect
- The effect becomes less prominent as the electronegativity of X increases
- This is because electronegative atoms have a higher effective nuclear charge, which causes its HOMO to be lower in energy. As a result, the gap between the HOMO of the substituent X and the π* orbital of C=O to be larger, which decreases the effectiveness of the overlapping
The lone pair electrons on the carbonyl oxygen is delocalized with the σ* orbital of C–X
- This makes the oxygen a weaker nucleophile
The reactivity of carboxylic acid derivatives can be explained by the above two molecular orbital interactions
- The lone pair on the substituent atom becomes less nucleophilic and the carbonyl carbon becomes less electrophilic as the donation of lone pair from the substituent to the π* orbital of C=O becomes stronger
The interaction both lowers the energy of the energy of the substituent's lone pair and raises the energy of the π* orbital
- The lone pair is stabilized, making it less basic and nucleophilic
- The C=O π* goes up in energy, making it less ready to react with nucleophile. In addition, the
- The nucleophilicity of the lone pair and the electrophilicity of the acyl carbon decreases as the electronegativity of the attached substituent decreases
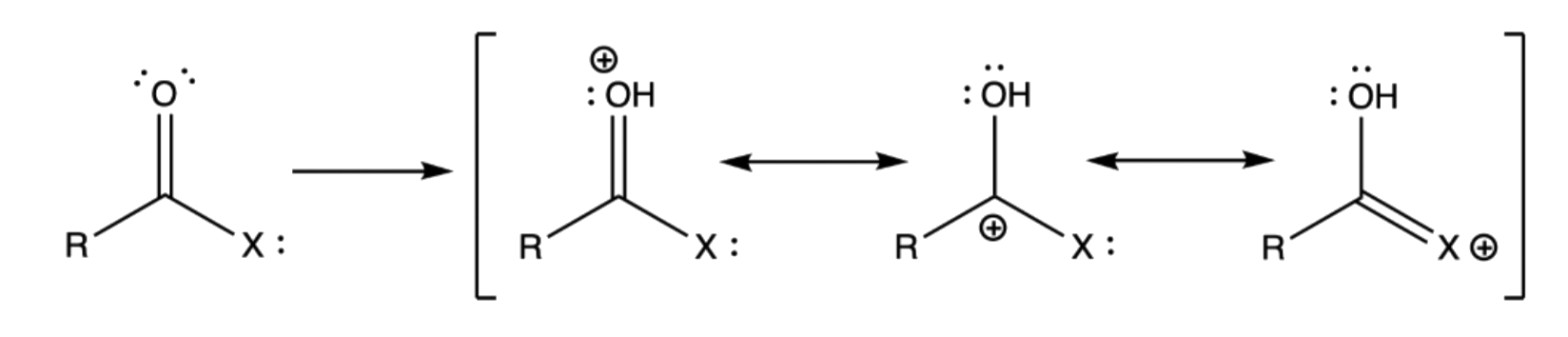
Acidity and Basicity of Carboxylic Acid Derivatives
Carboxylic acid derivatives are basic and acidic
Protonation always occurs on the acyl oxygen, and not on the substituent
- The lone pair on the substituent cannot be protonated because it is delocalized and stabilized, which makes it much lower in energy than the lone pairs of acyl oxygen
- In all cases, protonation requires a strong acid, but it gets easier as the electron-donating ability of the substituent increases, which is demonstrated in the resonance structures
- The resonance structure where the substituent is bearing the positive charge is a relatively strong contributor to the overall resonance hybrid
- The protonated form of these derivatives can be further stabilized by increasing the electron-donating effect of the substituent
- An easier way to rationalize this is that by decreasing electronegativity of the substituent, it will be more capable of bearing the positive charge, thus stabilizing the structure
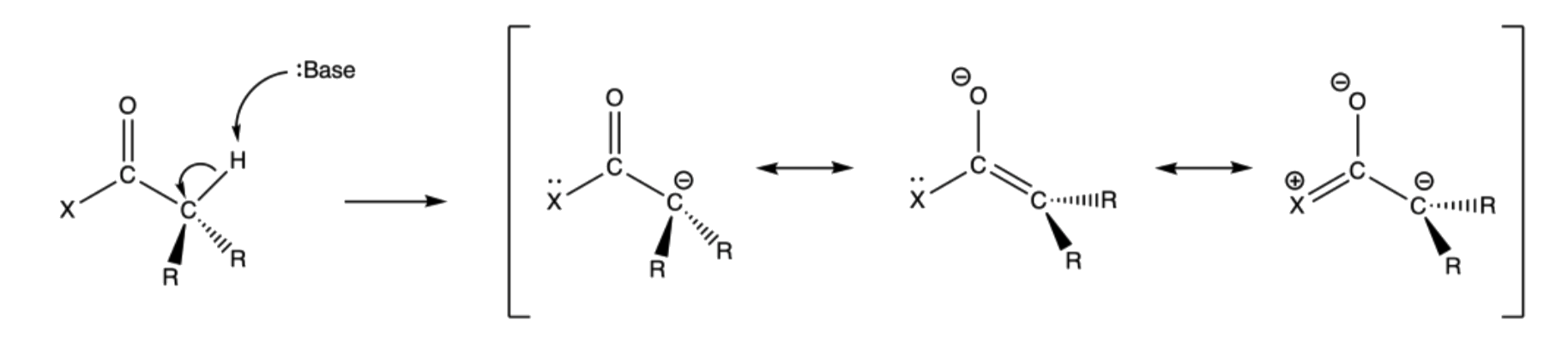
Just like ketones and aldehydes, the alpha hydrogen of carboxylic acid derivatives is also acidic
- The alpha hydrogen of carboxylic acid derivatives is less acidic than their carbonyl counterpart
- Just like ketones and aldehydes, the conjugate base of the carboxylic acid derivatives is stabilized by resonance
- Unlike the ketones and aldehydes, there is also a resonance structure where the substituent is donating electron density to the carbonyl carbon, which competes with the stabilization of the enolate charge
- Due to the extra unstable resonance structure, the conjugate base of carboxylic acid derivatives is less stable than those of aldehydes and ketones, which makes them less acidic
- The acidity of the alpha hydrogens of carboxylic acid derivatives increases as the substituent becomes more electronegative
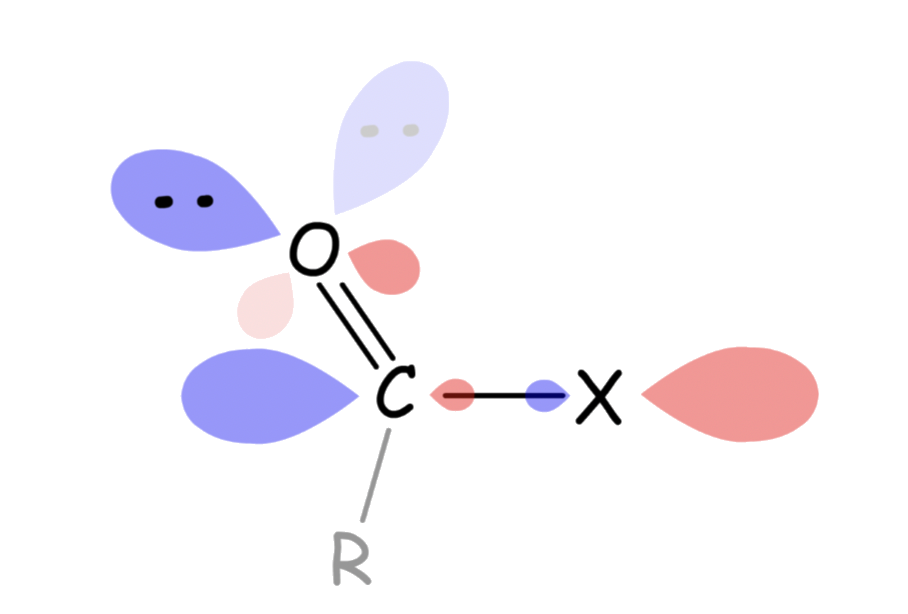
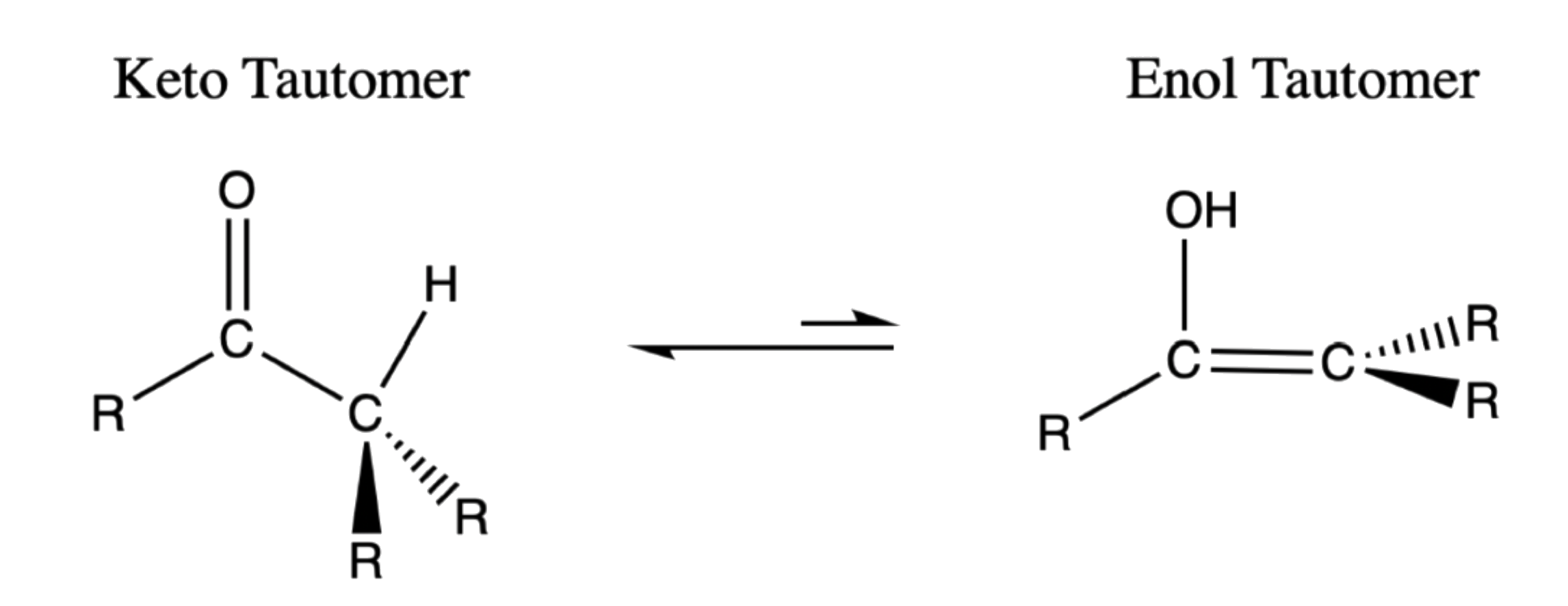
¶ Enol
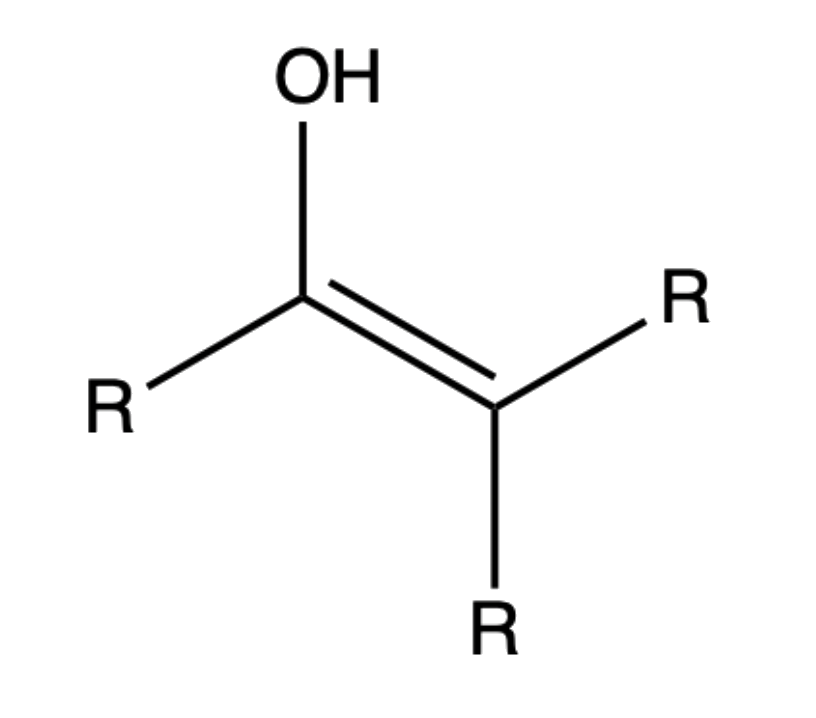
Enols, or more formally, alkenols, are a type of structure that is represented as an alkene with a hydroxyl group attached to one end of the alkene double bond
¶
Keto-enol Tautomerism
Because of the acidity of α hydrogens, carbonyls and carboxylic acid derivatives undergo keto-enol tautomerism
Tautomers are rapidly interconverted constitutional isomers, usually distinguished by a different bonding location for a labile hydrogen atom and a differently located double bond
- The equilibrium between tautomers is not only rapid under normal conditions, but it often strongly favors one of the isomers
The formation of enols can be catalysed by acid or base
¶ Ionic and Radical Carbons
¶ Carbocation
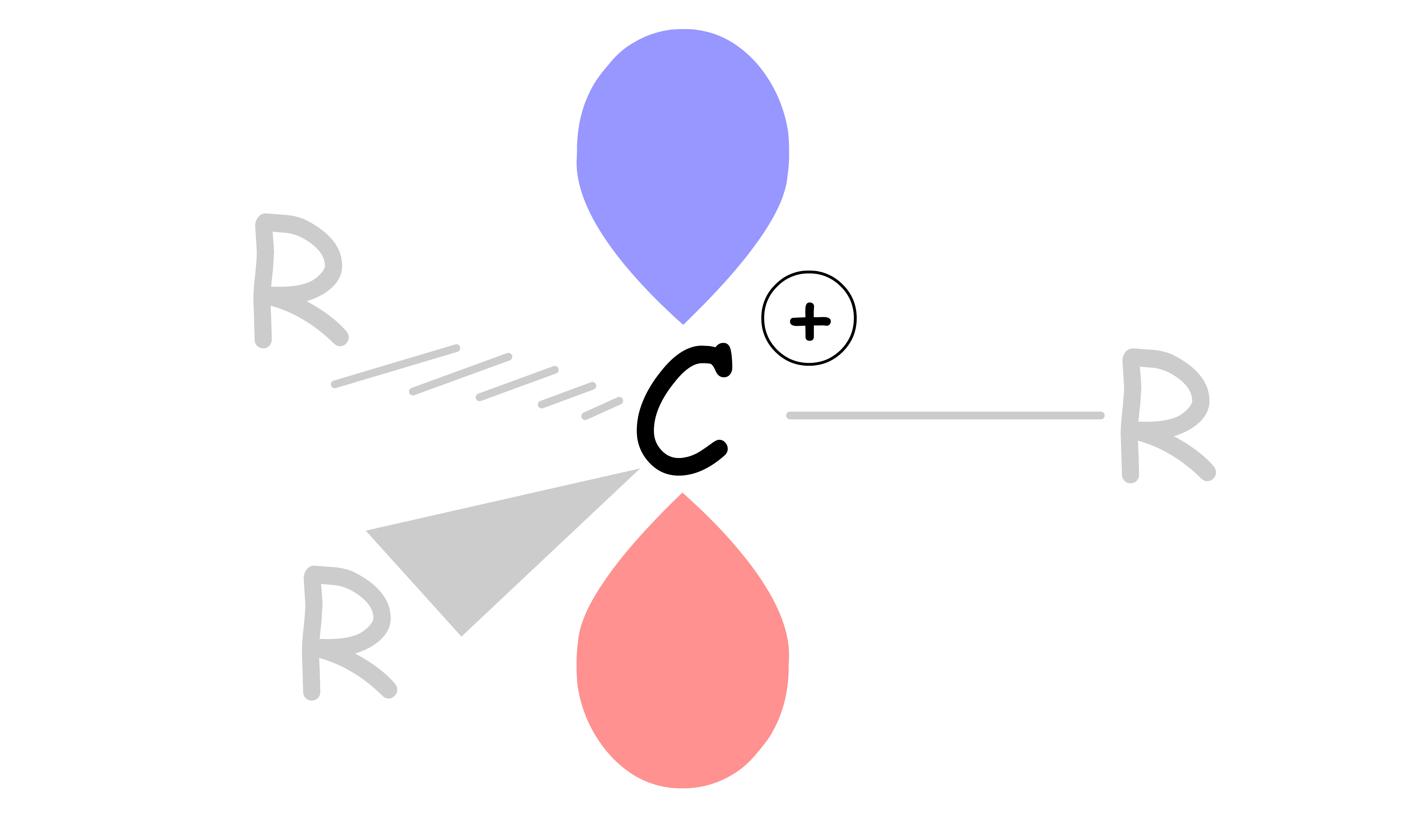
A carbocation is an ion with a positively-charged carbon
- There are only 6 valence electrons on the carbon instead of obeying the octet
- All 6 electrons are used to form 3 sigma bonds, leaving an empty p orbital extending above and below the plane occupied
Carbocations are often reactive, seeking to fill the octet of valence electrons as well as regain a neutral charge
Carbocations are planar, with the trivalent carbon being sp2 hybridized
¶
Stability of carbocations
There are three factors that contribute to the stability of carbocations
- The inductive effect
- The effect of the sigma electron displacement through sigma bonds
- Carbocations are very electron-poor, and thus anything which donates electron density to the center of electron poverty will help to stabilize it
- Attaching Electron-Donating Groups to the carbocation helps stabilizes it
- The higher the degree of substitution, the stronger the stabilizing inductive effect
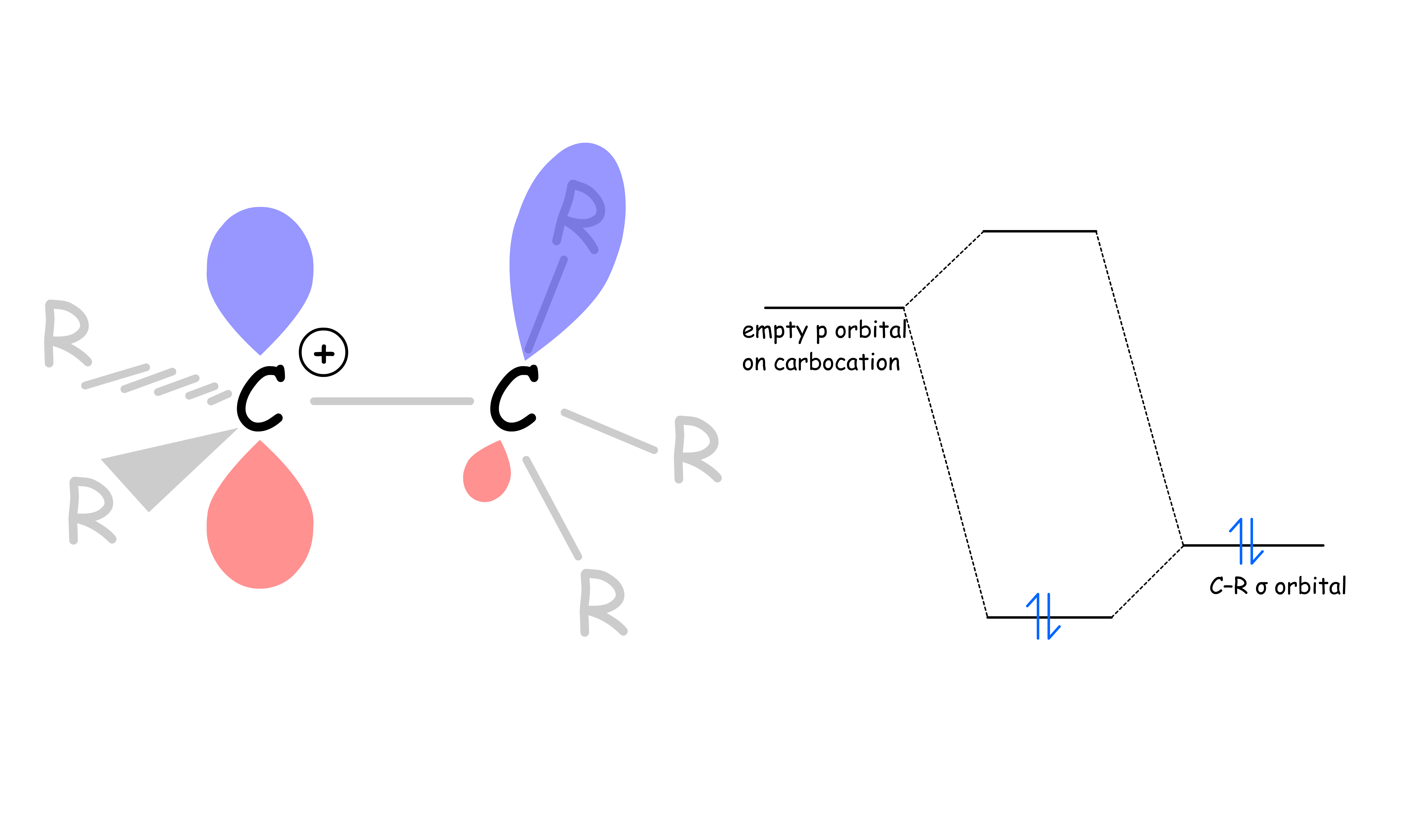
- Sigma conjugation
- Electron delocalization could also occur via parallel overlap of p orbitals with hybridized orbitals participating in sigma bonds
- Sigma conjugation involves partial mixing of a sigma-bonding orbital of an adjacent C–R bond with the vacant 2p orbital of the carbocation
- Sigma conjugation occurs between the empty p orbital of the carbocation and the properly oriented sigma bonds on neighboring carbons that are roughly parallel to the p orbital
- The higher the degree of substitution, the more possibilities there are for sigma conjugation, and the more stable the carbocation
- Resonance
- Resonance is a much stronger stabilizing effect than the other two
- The positive charge is not localized in the carbocation, rather it is spread throughout the site of delocalization
- Benzylic and allylic carbocations (where the positively charged carbon is conjugated to one or more non-aromatic double bonds) are very stable due to the high degree of delocalization
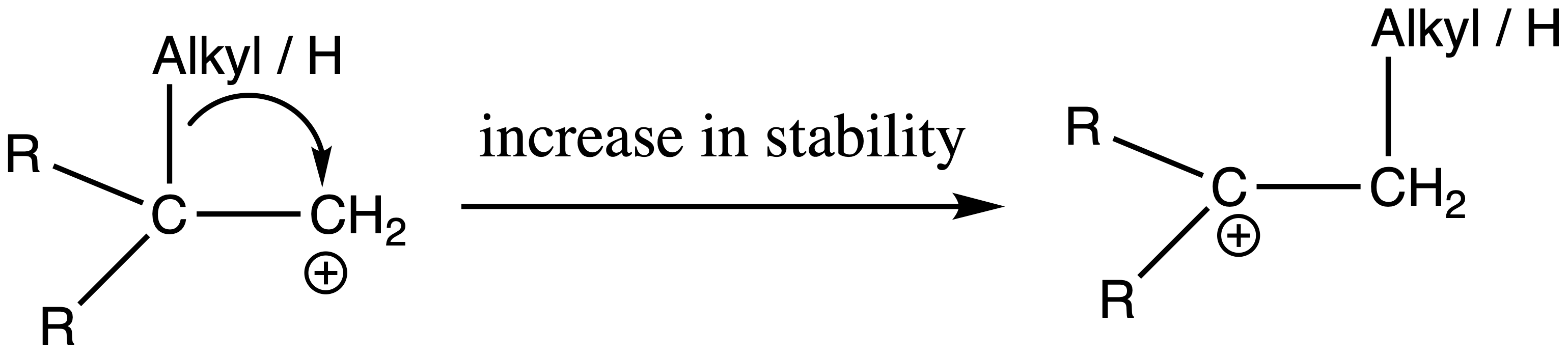
Carbocation Rearrangement
Carbocation are defined as the movement of a carbocation from an unstable state to a more stable state through the use of various structural reorganizational "shifts" within the molecule
- The molecule will transforms from one unstable carbocation to a carbocation that is relatively more stable
There are different types of carbocation rearrangement
- Typically, either an alkyl group or a hydrogen migrates, each with its bonding electrons, from an adjacent atom to the electron deficient carbocation
- A less stable carbocation rearranges to a more stable ion ( from a less substituted carbocation to a more substituted carbocation
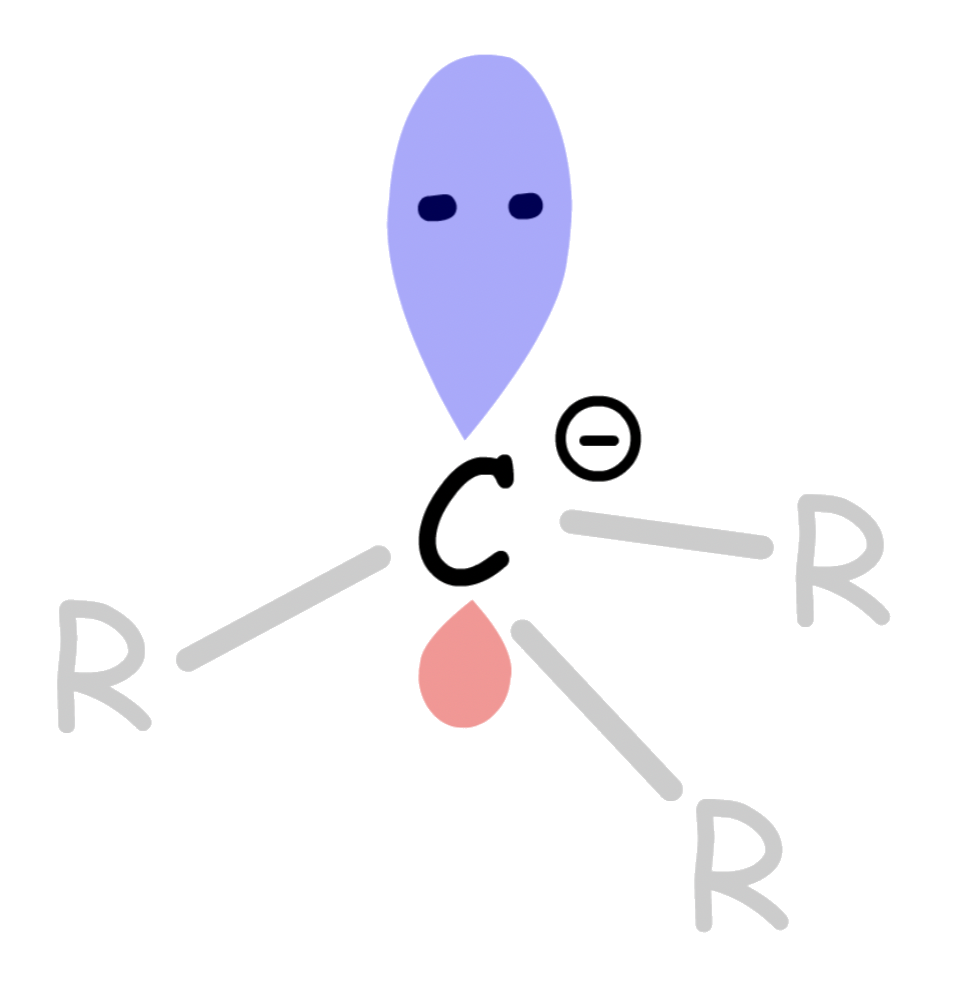
¶ Carbanion
A carbanion is an anion in which carbon has an unshared pair of electrons and bears a negative charge usually with three substituents for a total of eight valence electrons
Carbanion are tetrahedral, with the negatively charged carbon being sp3 hybridized
¶
Stability of carbanions
There are two main factors that contribute to the stability of carbanions
- The inductive effect
- The effect of the sigma electron displacement through sigma bonds
- Carbanions are very electron-rich, and thus anything which withdraw electron density away from the carbon will help to stabilize it
- Attaching Electron-Withdrawing Groups to the carbocation helps stabilizes it
- Hybridization of the charge-bearing carbon
- The lone pair of the carbanion is usually the HOMO
- The greater the s-character of the charge-bearing atom, the more stable the anion
- Usually carbanions are sp3 hybridized, but situations that causes the charge-bearing carbon to adopt hybridization with stronger s character will help stabilize the charge by pulling the HOMO down in energy
Chiral carbanions
With the molecular geometry for a carbanion described as a trigonal pyramid the question is whether or not carbanions can display chirality
- If the activation barrier for inversion of this geometry is too low any attempt at introducing chirality will end in racemization, similar to the nitrogen inversion
Evidence reveals that chiral carbanions indeed exist
- Chirality of carbanion is usually only relevant at low temperatures
- At high temperatures, the carbanions can easily undergo inversion and racemize themselves
¶ Carbon Radical
Radicals are atoms, molecules or ions with unpaired electrons that are highly reactive
- Some transition metals can have unpaired electrons, but they are relatively stable, so they are not considered as radicals
- Radicals can be neutral, positively or negatively charged
Stability of carbon radicals
Carbon radicals can be stabilized by an high degree of substitution
- The ranking of stability: 3° radical > 2° radical > 1° radical > Methyl radical
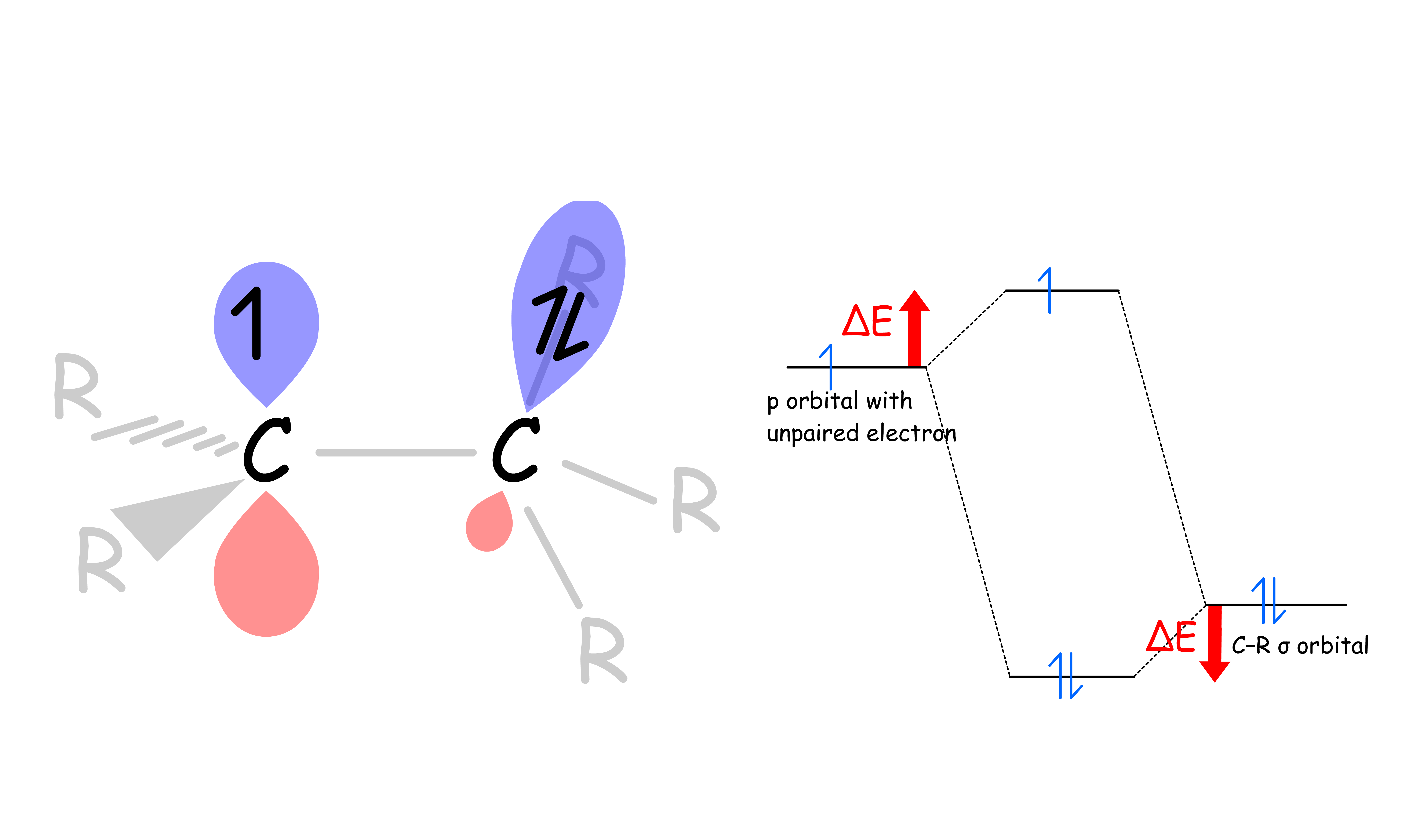
- The stabilization originates from the sigma-conjugation between the σ orbital of C–R and the SOMO ( Singly Occupied Molecular Orbital )
- The radicals are ( usually ) sp2 hybridized with an orthogonal p orbital containing the unpaired electron. Neighboring C–R bonds can overlap with the p orbital to form 2 new molecular orbitals
- The two electrons in the C–R sigma bond are stabilized by ΔE, while the unpaired electron in the SOMO is destabilized by ΔE
The overall effect is stabilizing increases by a factor of ΔE for a higher degree of substitution
Radicals can also be stabilized through resonance
Resonance is possible if neighboring atoms have an p orbital that is parallel to the SOMO p-orbital of the radical