An electrophilic addition reaction is a reaction in which a substrate is initially attacked by an electrophile, and the overall result is the addition of one or more relatively simple molecules across multiple bonds
- The reactions shown below occur in the same way for both alkene and alkyne
The regioselectivity of these reactions can be described by using the Markovnikov's rule
- The rule state that for the electrophilic addition of an asymmetric alkene, the electrophile will be attached to the less substituted carbon, while the second species will be attached to the more substituted carbon
- For reactions that give products that are contrary to this rule ( i.e. the electrophile is attached to the more substituted carbon, while the second species will be attached to the less substituted carbon ), we say they are Anti-Markovnikov's reactions
Although addition reaction is not strong enough to disrupt the aromaticity of compounds, it does not mean aromatic compounds cannot undergo addition reactions
- Addition reaction does not happen to the conjugated p-orbitals
- If a triple bond is present in the aromatic compound ( aryne ), then addition reaction can occur on that set of p-orbitals as it is not stabilized
¶ Acidic Ionic Addition
The acidic ionic addition requires the presence of hydrogen ions, the nucleophile
- The nucleophile ( ) can be halides, water, sulphate ion etc.
A solution often contains more than one nucleophile, so the major product is formed by the forming bonds with the stronger nucleophile
- Even if the weaker nucleophile manage to form the single bond with carbon, it can still be easily displaced through substitution reaction by a stronger nucleophile
¶ Mechanism
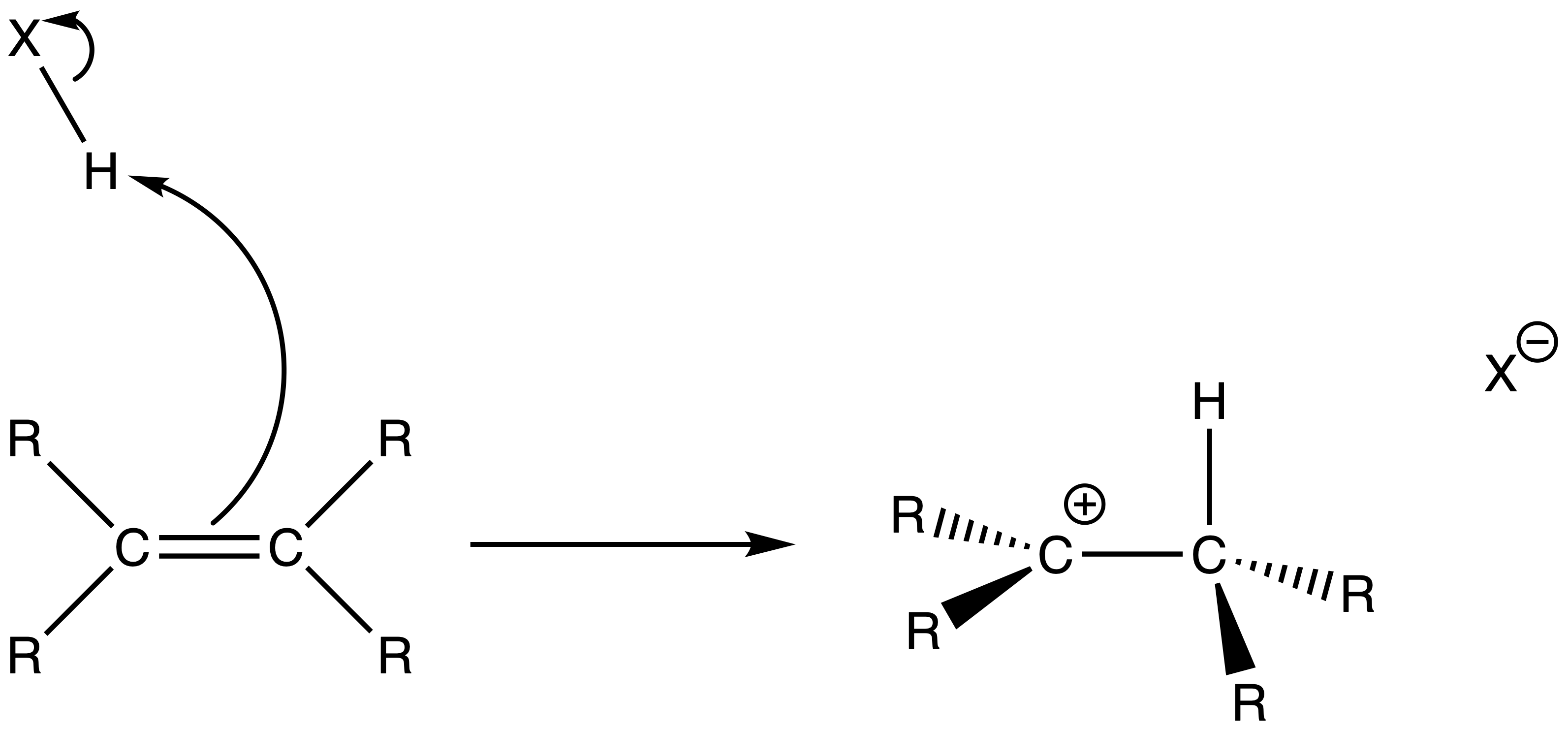
- The reaction begins when the alkene nucleophile donates a pair of π electrons from the C=C bond to HX to form a new C–H bond
- A carbocation intermediate is formed
- The carbocation is preferred to form on the more substituted carbon due to a better sigma-conjugation
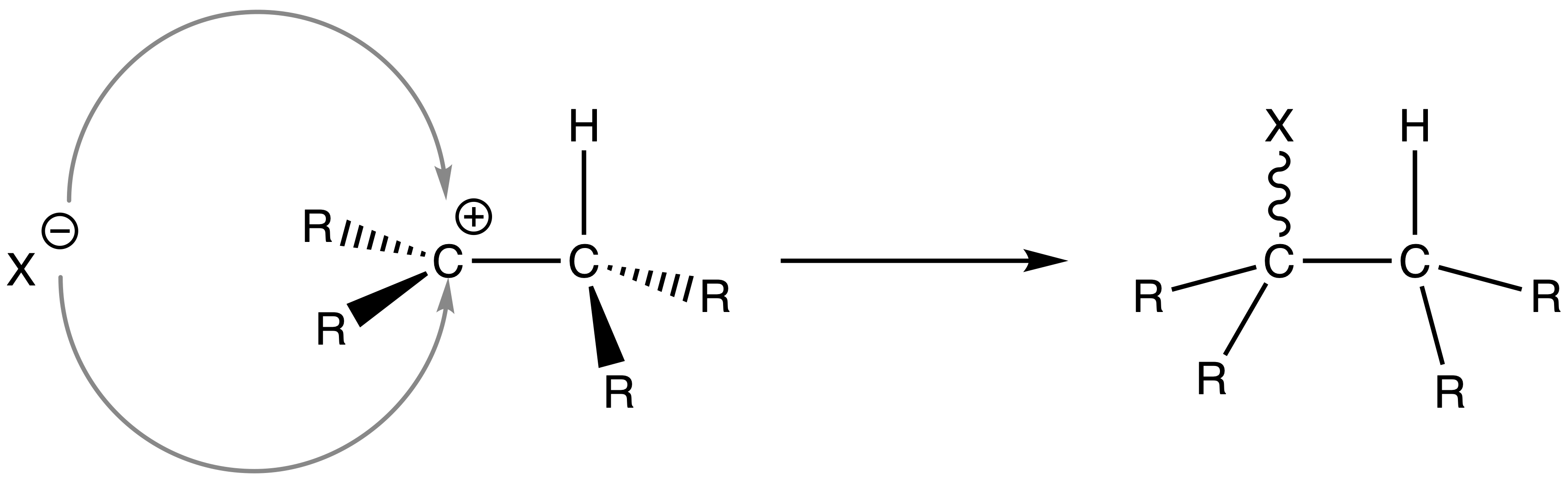
- The nucleophile donates an electron pair to the vacant p orbital of the carbocation, forming a C–X bond and yielding the neutral addition product
- The nucleophile can attack from either the si face or the re face
- The product will be a mixture of enantiomers / diastereomers
¶ Stereochemistry and Regiochemistry
The reaction is regioselective
- The product can be predicted using the Markovnikov's Rule
- The regioselectivity of the reaction originates from the stability of carbocation
- The formation of carbocation is the rate-determining step, so structures that can stabilize the carbocation to the higher degree will be favored
The reaction is not stereoselective
- The nucleophile can attack from either side of the carbocation intermediate, thus forming a racemic mixture
¶ Anti-Addition via a Three-Membered Ring Pathway
Addition reaction can proceed through the formation of a three-membered ring intermediate
- There are three main three-membered rings that can act as the intermediate
- Bridged halonium ion
- Bridged mercurinium ion
- Epoxide
The addition reaction involves formation of a three-membered ring followed by an anti-attack by a nucleophile
- Despite their similarities, the three three-membered rings are quite different in origin and properties
¶ Formation of the Three-membered ring
Bridged Halonium Ion
A halogen bearing a positive charge is called a halonium ion, and the cyclic structure of which it is a part is called a bridged halonium ion ( R2X+ )
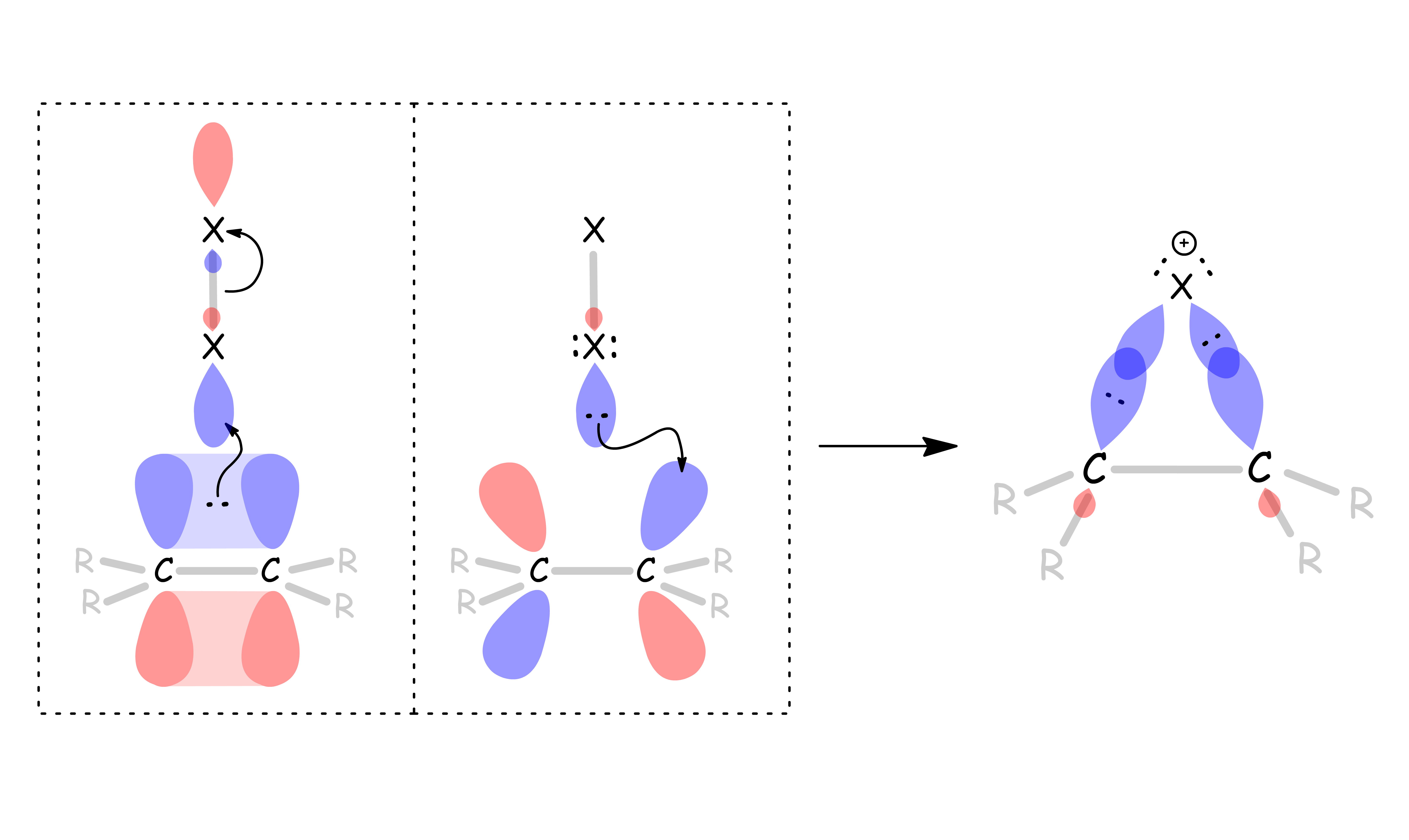
Formation of a bridged halonium ion :
- The π orbital of the alkene overlap with the σ* orbital of the halogen, thus forming a C–X bond and breaking a X–X bond simultaneously
- A lone pair on the halogen is donated to the π* orbital of the alkene
- As a result, the halogen forms two σ bonds with the carbon chain to give a cyclic ring, halonium ion
- In reality, the formation is more complex than that. The initial step is coordination of the halogen X2 in a loosely bonded π complex. The complex then breaks down to give the halonium ion
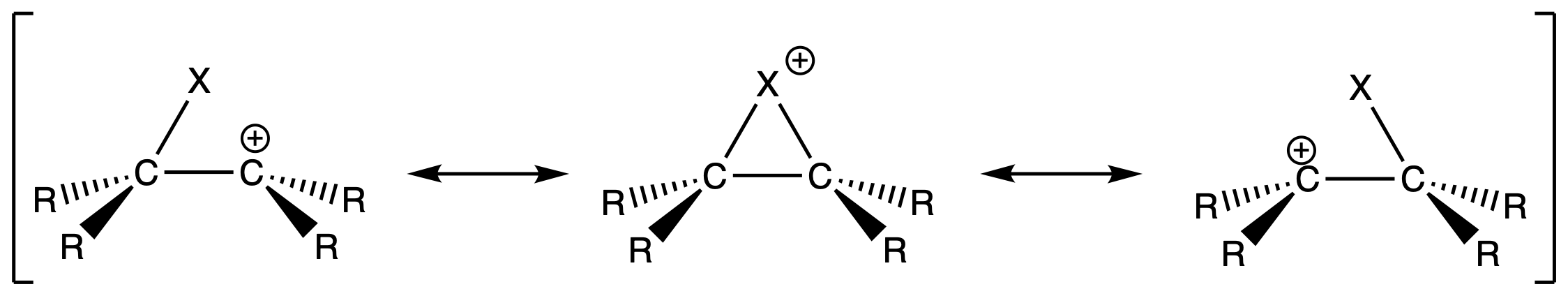
The bridged halonium ion has multiple resonance structures
- Halogens are electronegative atoms, so they cannot bear the positive charge very well
- The carbocations are the major contributor to the delocalized structure
- The bridged halonium is unstable, but its existence defines the geometry of the delocalized structure
- Because of the planar geometry of alkene, the bridged halonium ion can form with equal probability on the top or bottom face of the alkene
Bridged Mercurinium Ion
The bridged mercurinium ion is an extremely important intermediate as no discrete carbocation intermediate
- Complication caused by carbocation rearrangement can be avoided
Formation of the mercurinium ion :
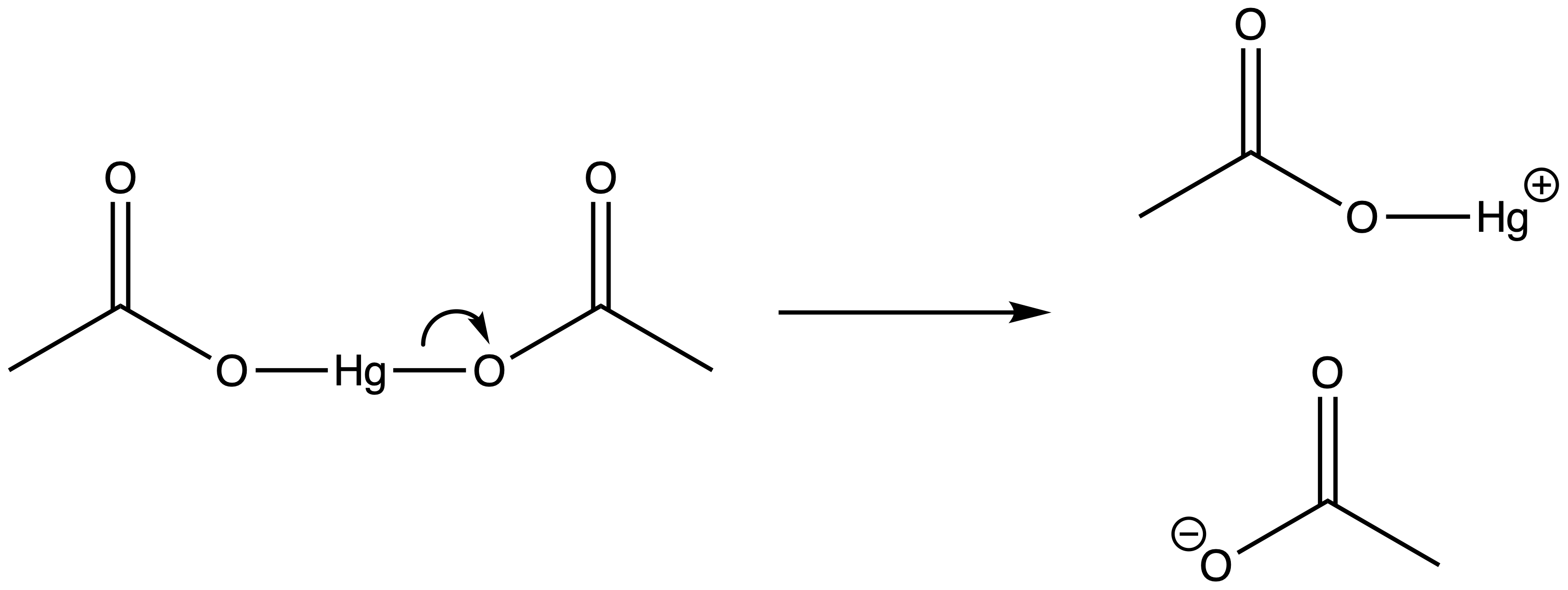
- Dissociation of mercuric acetate gives AcOHg+ ( an electrophile ) and acetate ion
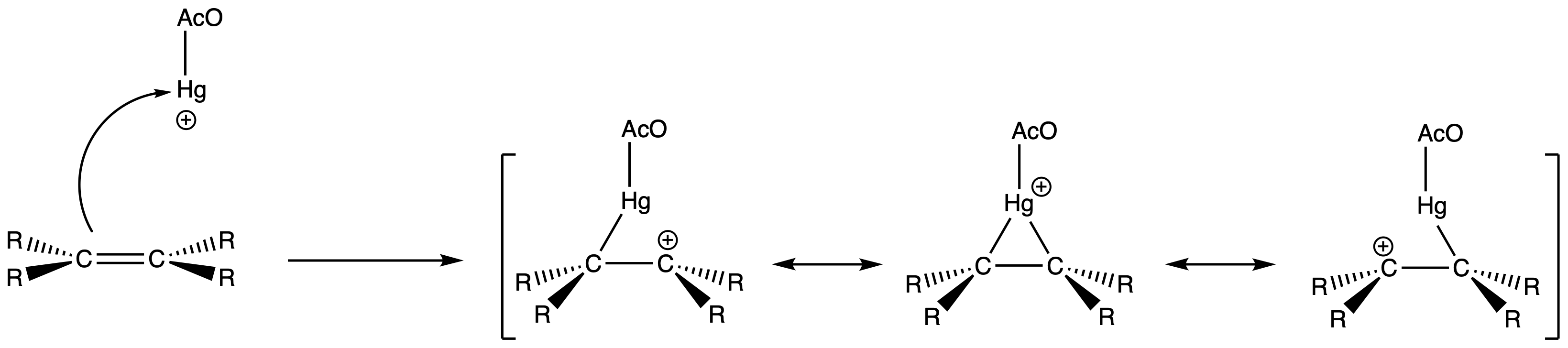
- Attack of on the π bond forms a bridged mercurinium ion intermediate
¶
The two π electrons from the carbon-carbon double bond form a ring containing three atoms
- The ring is a two electron-three center system
The contribution of the open carbocation intermediate to the resonance hybrid decreases as their respective degree of substitution decreases
- Their ability to stabilize the carbocation decreases with the degree of substitution
The fact that oxymercuration occurs without rearrangement indicates that the intermediate formed in this step is not a true carbocation
- It is instead a resonance hybrid largely with the character of a bridged mercurinium ion
Note that the mercury acetate is usually reduced by NaBH4 after the anti-addition of the nucleophile
- Reduction of C–HgOAc bond to a C–H bond to give the final product and metallic mercury
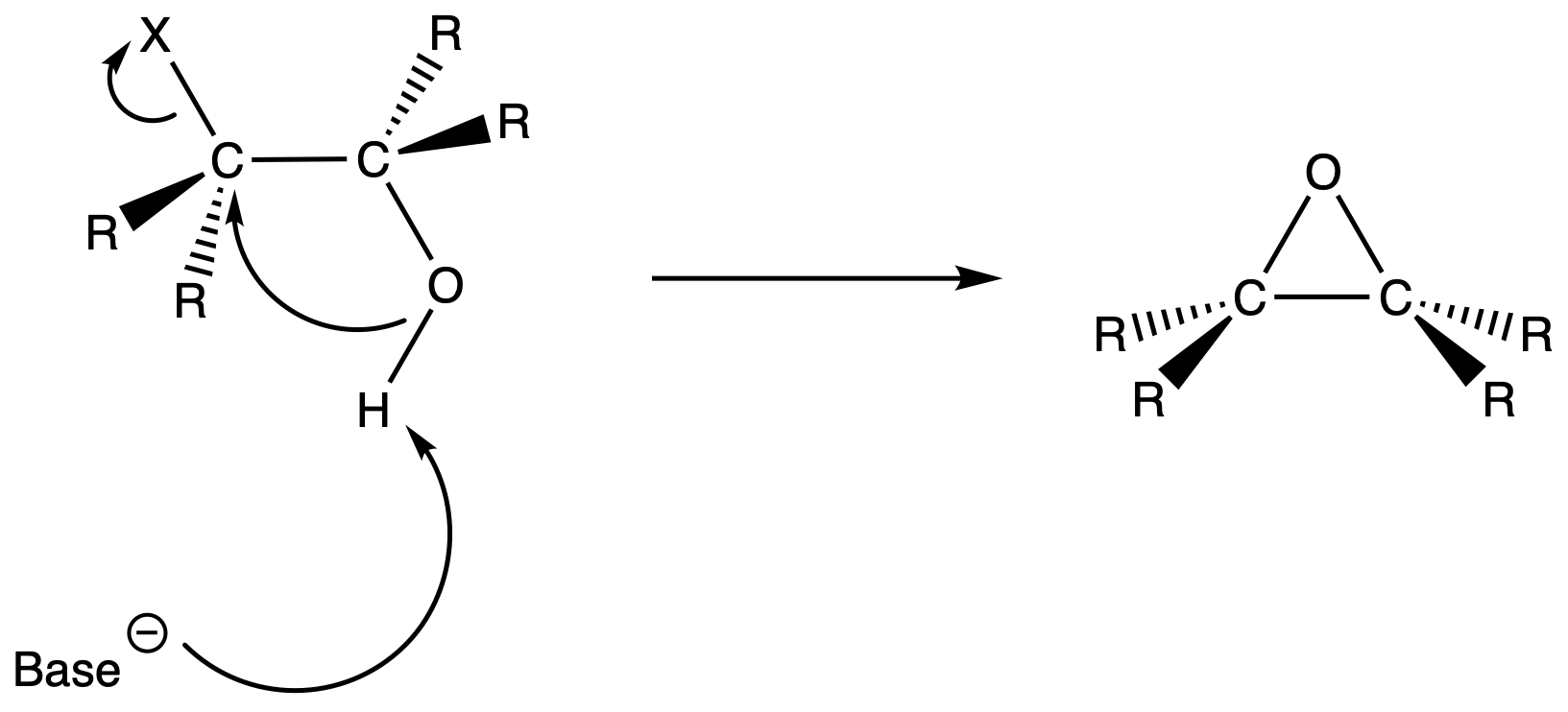
Epoxides
Epoxides are three-membered rings that contain an oxygen atom
- Epoxide is a very reactive intermediate, but also stable enough to be isolated
- Epoxide is much more reactive than ether due to its ring strain
There are two ways to form epoxide from an alkene
- Syn-addition using peroxyacid
- Formation of halohydrin followed by an elimination
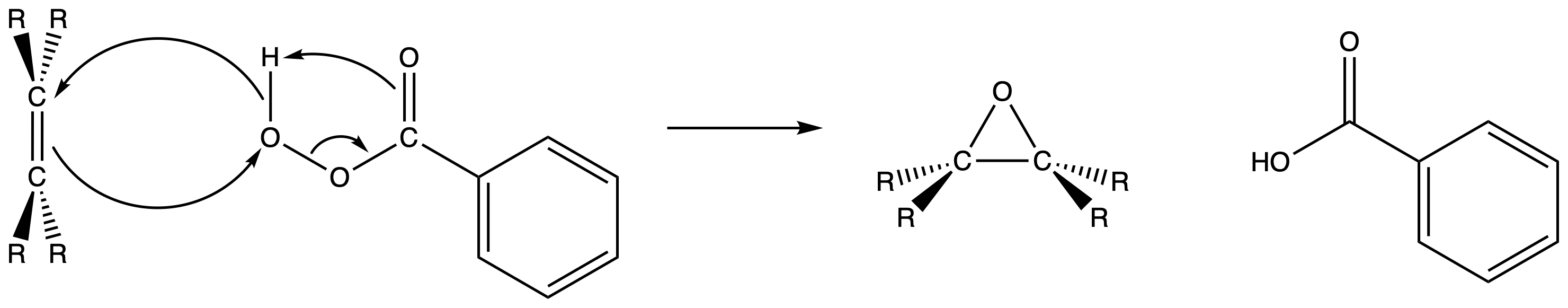
Syn-addition using peroxyacid
- The alkene react with the peroxyacid to form epoxide through a concerted mechanism
- The peroxyacid transfer an oxygen atom to the alkene with synchemistry
- Both C–O bonds form on the same face of the double bond through a one-step mechanism with no intermediates
The oxygen atom farthest from the carbonyl group is the one transferred
- More electron-rich alkenes are epoxidized faster, so alkene with a higher degree of substitution will have a faster rate
- The oxygen that is added to the carbon chain is a strong electrophile
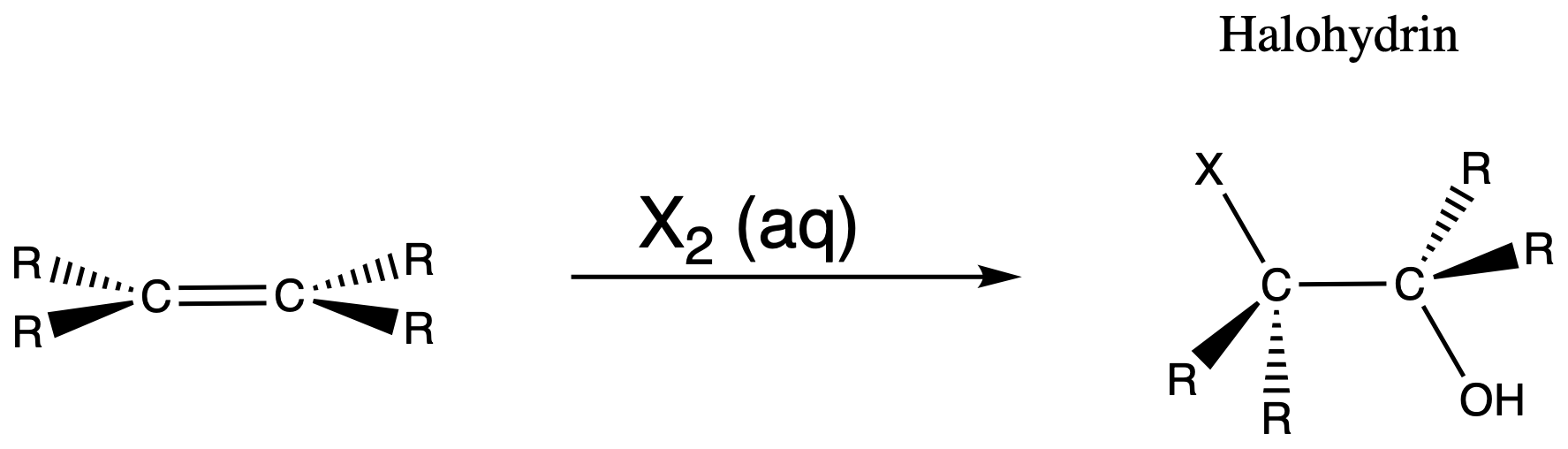
Formation of halohydrin followed by an elimination
- Formation of halohydrin by treating the alkene with a halogen in aqueous solution (you should understand why this reaction happen after reading this section)
- Treat the halohydrin with base to eliminate HX and produce an epoxide
¶ Stereochemistry and Regiochemistry
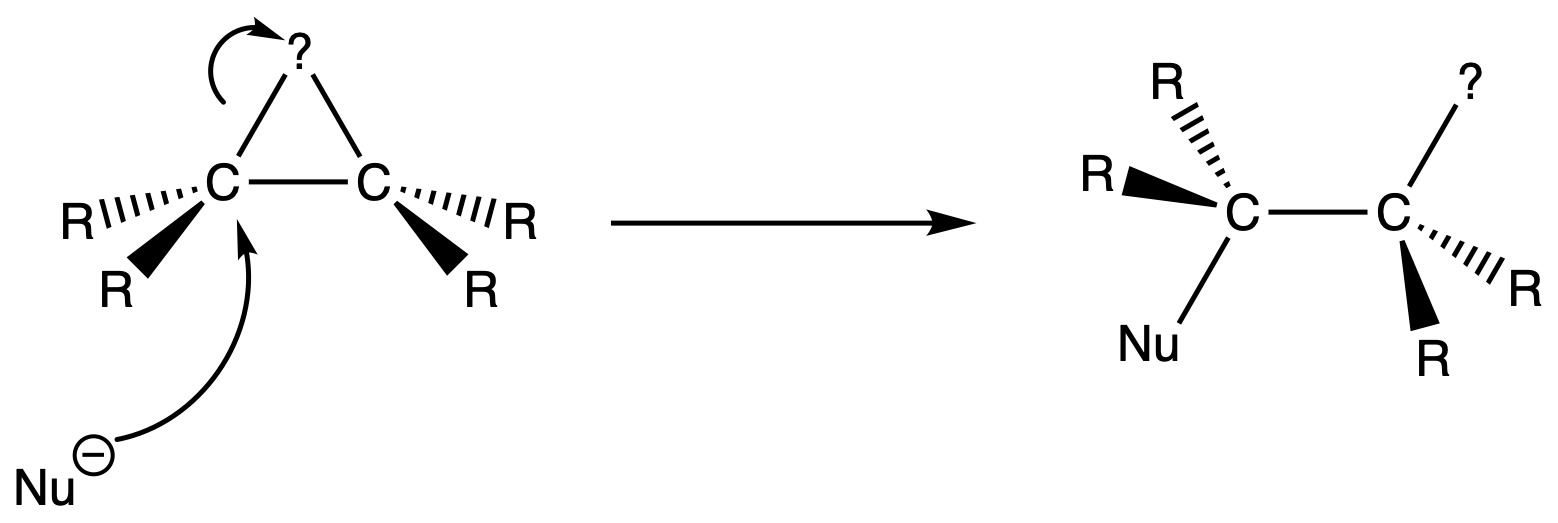
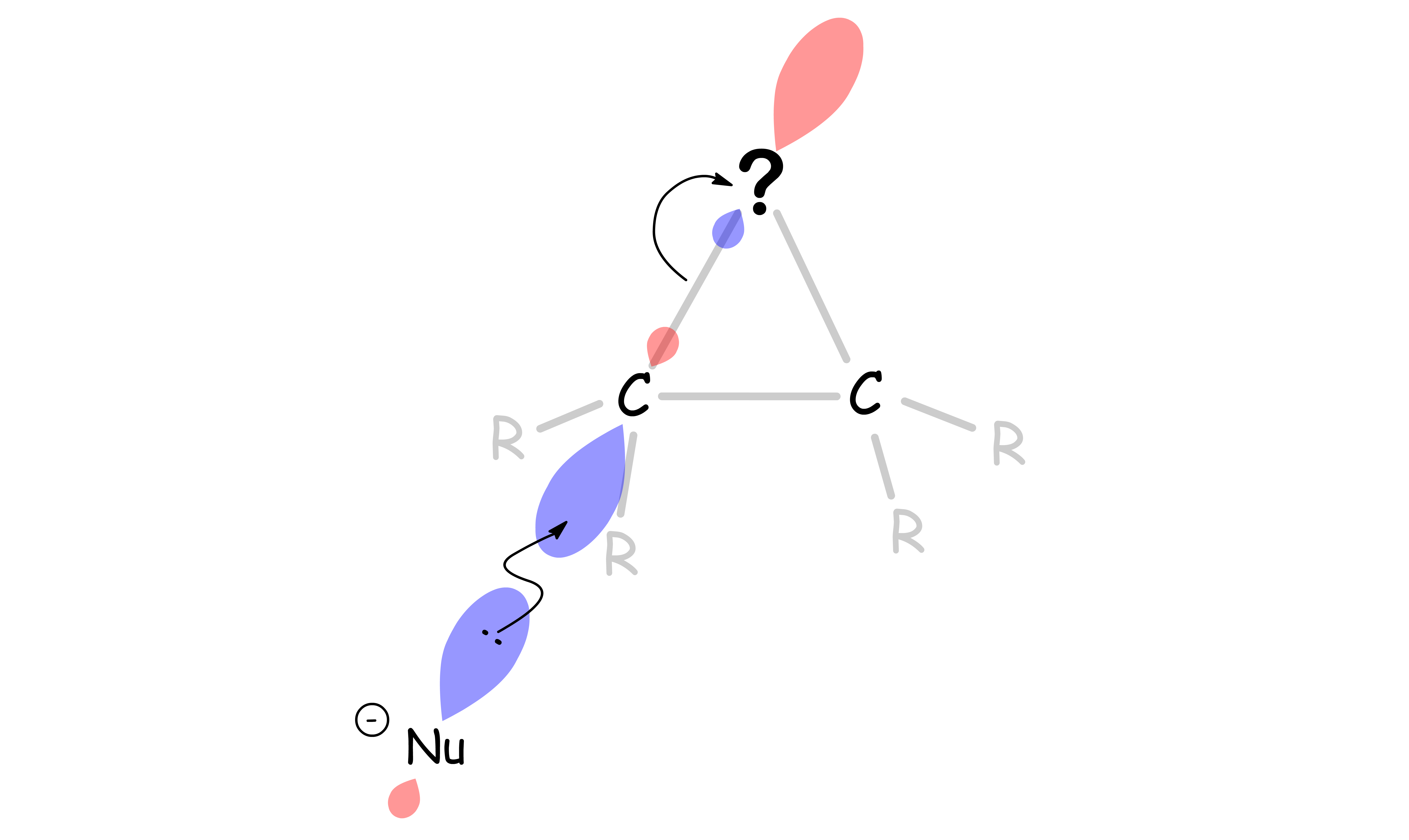
The addition of the nucleophile is stereospecific
- Attack of nucleophile on carbon from the side opposite to the ring
- The three-membered ring is opened during the process
- The heteroatom in the three-membered ring and the nucleophile are not only added in an anti-fashion, but also in the same plane ( anti-periplanar )
- We call this an anti-coplanar attack
Note that the addition is not stereoselective, but stereospecific
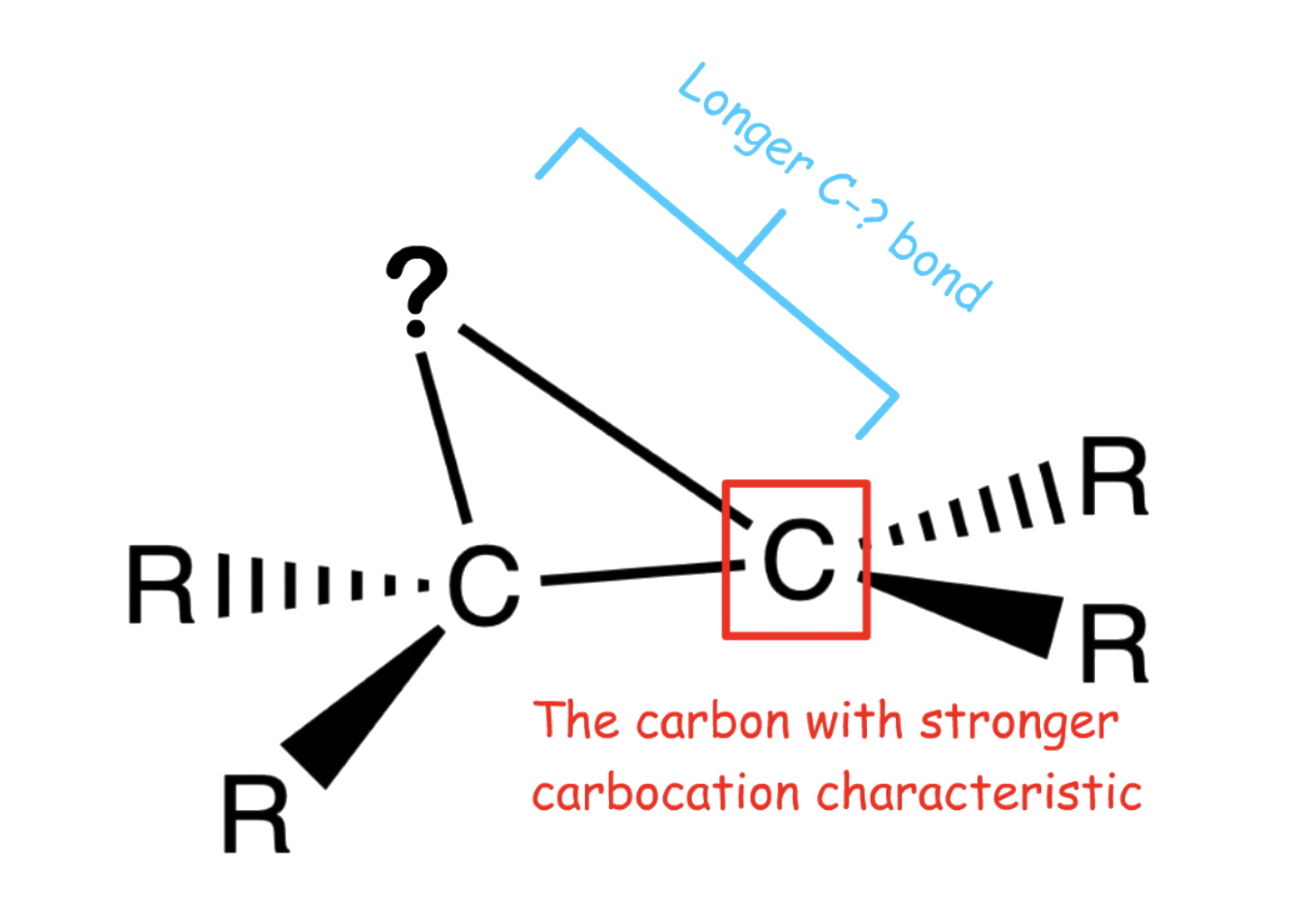
- The nucleophile can only attack any carbon from the opposite side of the ring due to the position of the C–? σ* orbital
- It is an coplanar attack and an anti-addition since the heteroatom in the three-membered ring and the nucleophile are not only added on opposite side ( anti-periplanar ), but they also lie on the same plane
The reaction is regioselective
- The nucleophile prefers to attack the more substituted carbon
- There are two reasons that can explain the regioselectivity:
1. Carbocation character
- There is more carbocation character on the more substituted carbon, which directs attack of the nucleophile preferentially to that carbon
2. Steric hinderance
- The C–? bond to the more substituted carbon is longer ( weaker ) than its counterpart
- The longer C–? bond means that there is a larger space ( smaller steric hinderance ) for the nucleophile to attack
¶ Addition through Hydroboration
Hydroboration refers to the addition of a hydrogen-boron bond to C-C, C-N, and C-O double bonds, as well as C-C triple bonds
- Important for cross coupling reactions
- The organoborane molecule produced in the reaction can react with a variety of reagents
Hydroboration differs from many alkene addition reaction in that it occurs in a single step without a carbocation intermediate
- This method avoids complication of carbocation rearrangement
Borane CANNOT be prepared as a pure compound as it dimerizes to diborane, B2H6, a toxic gas that ignites spontaneously in air
- However, BH3 forms stable Lewis adduct with ethers
-Borane is most often used as a commercially available solution of BH3 in THF
¶ Mechanism
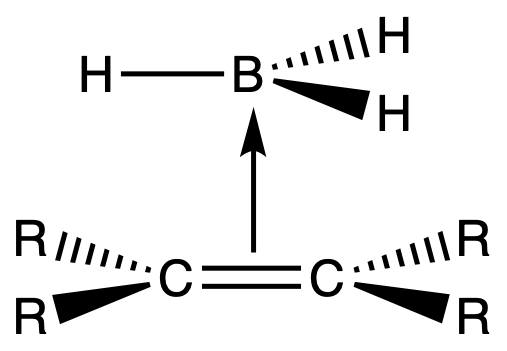
- The addition of borane to an alkene is initiated by coordination of the vacant 2p orbital of boron ( an electrophile ) with the electron pair of the π bond ( a nucleophile )
- The coordination is a Lewis acid-base interaction
- The borane lines up parallelly with the plane of the alkene
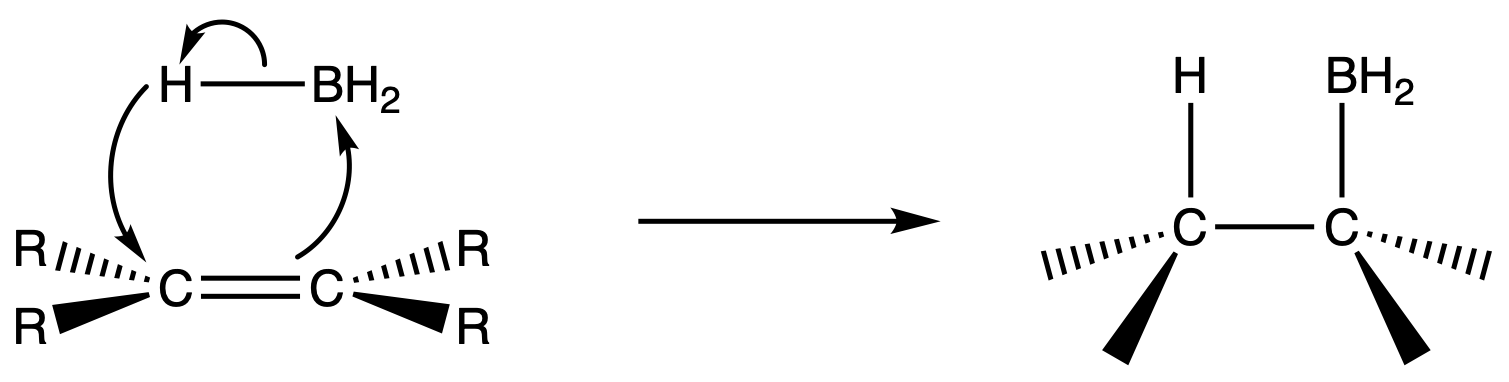
- The boron adds to the less substituted carbon of the alkene, thereby placing the hydrogen on the more substituted carbon, via a cyclic, four-centered transition state
- This is step involves a concerted six electrons movement
- Since everything happened in a single step, it is a syn addition
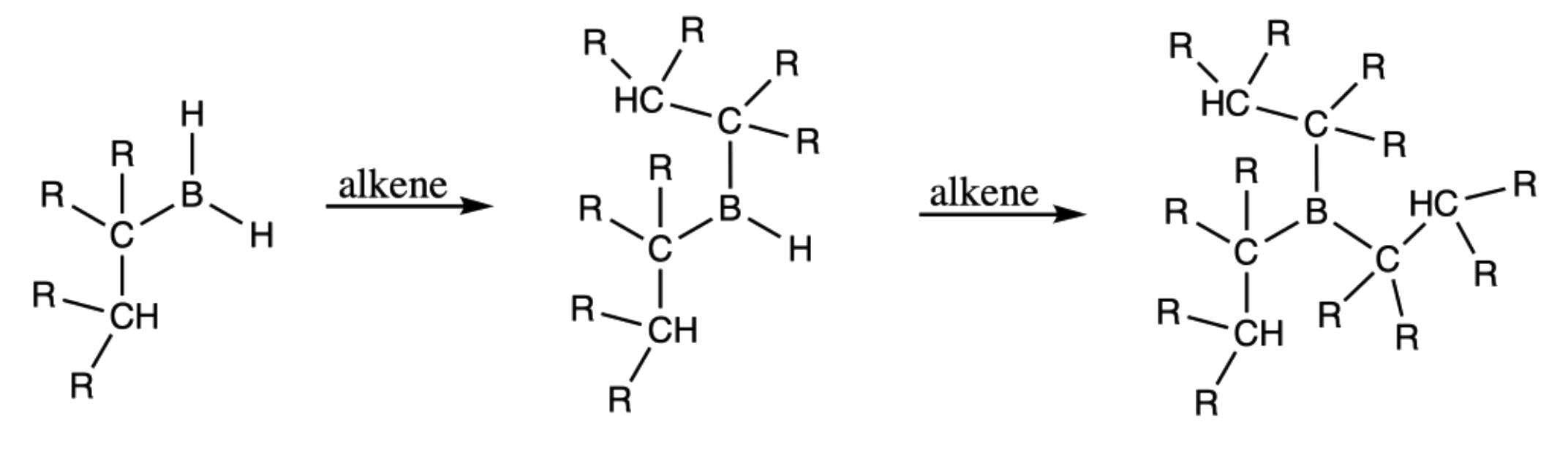
- Step 2 is repeated two more times to form the trialkylborane
- It is obvious that expressing the structure as such is fairly distracting, hence we can simply the structure as such:
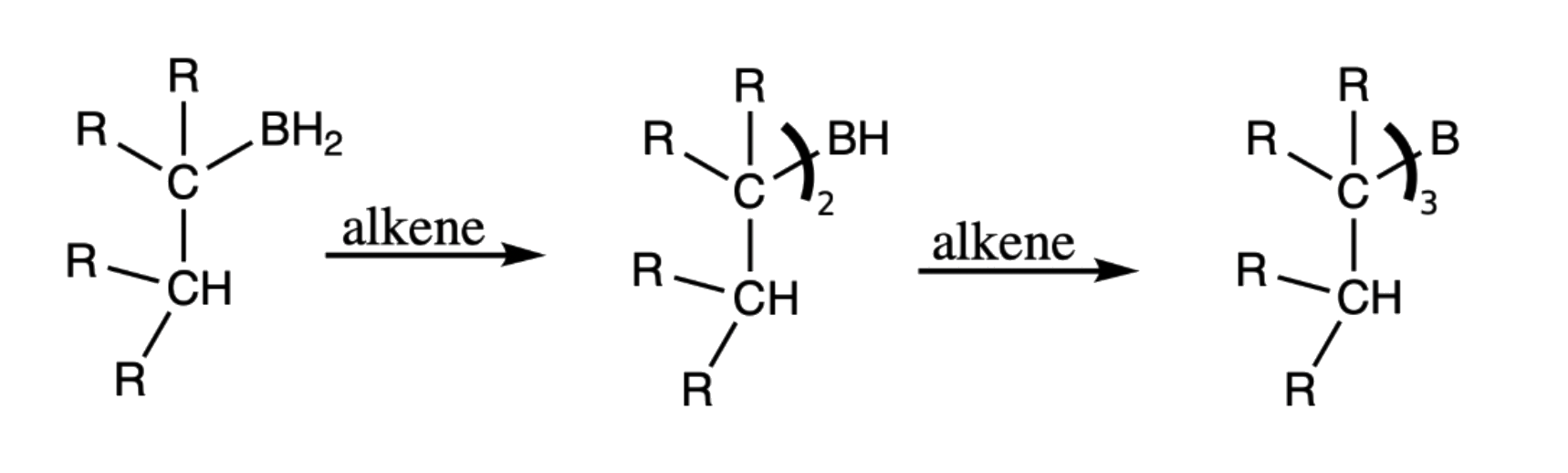
¶ Addition of Water
One of the most useful application of hydroboration is to add a hydroxyl group to an alkene
- This is useful because the hydroxyl group is added to the less substituted carbon, which is something most reactions cannot do
Mechanism:
- Hydrogen peroxide ion generates hydroperoxide ion in equilibrium in an alkaline medium
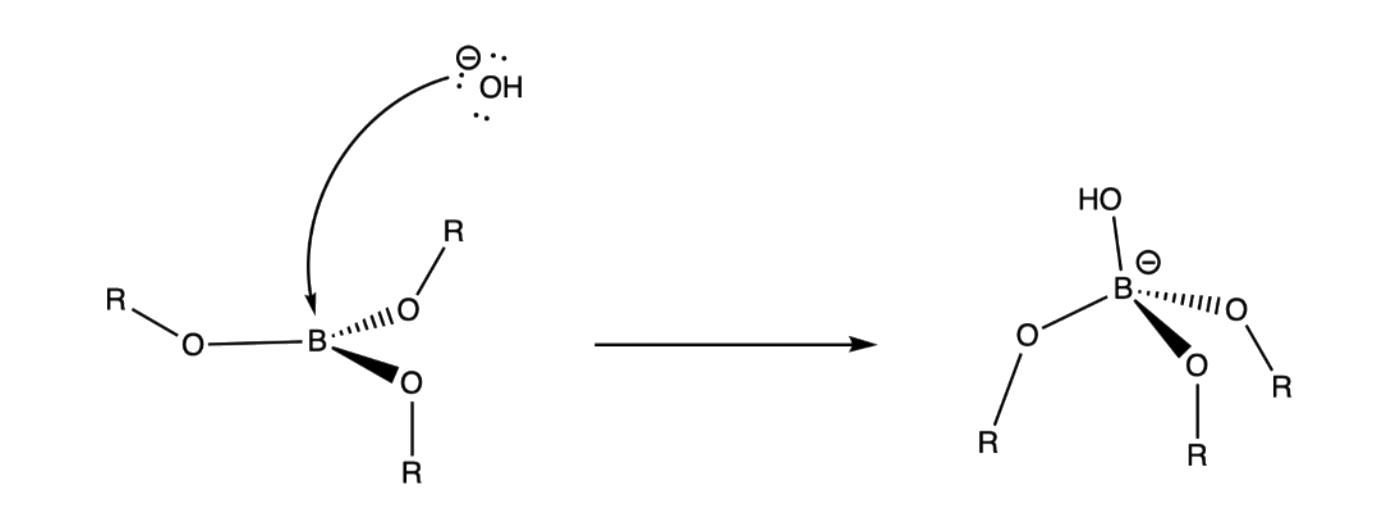
- The terminal oxygen of the hydroperoxide ion attacks the empty p-orbital of boron
3. Rearrangement of R group with its pair of bonding electrons to an adjacent oxygen results in ejection of hydroxide ion
- Boron has a formal negative charge, so it will want to eject a leaving group to regain its neutrality
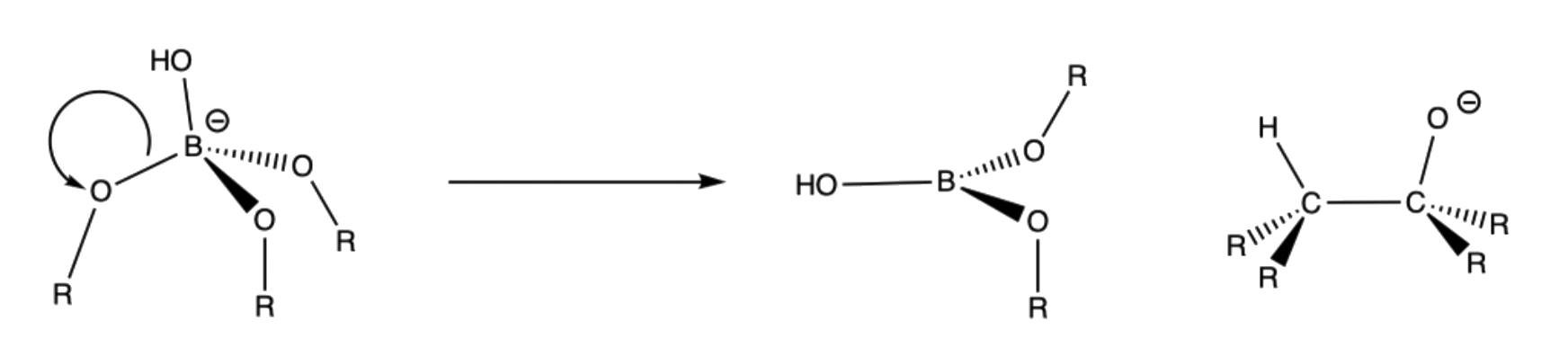
- B–O bond is favored over B–C bond because oxygen can stabilize the electron-deficient boron in a trigonal planar geometry through π-conjugation
- The R groups are poor leaving groups since the carbon stabilize the negative charge very well
- Due to all these electronic factors, it is thermodynamically stable to let the R group to migrate to the neighboring oxygen and eject a hydroxide group, which is a decent leaving group
- Migration occurs with retention of configuration of the migration group
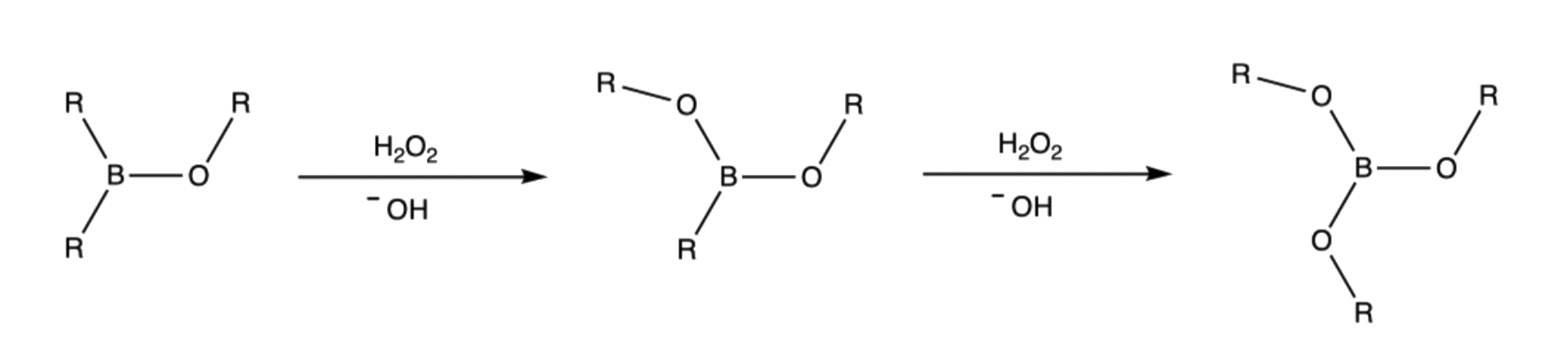
- Step 2 and step 3 are repeated two more time to form a trialkylborate
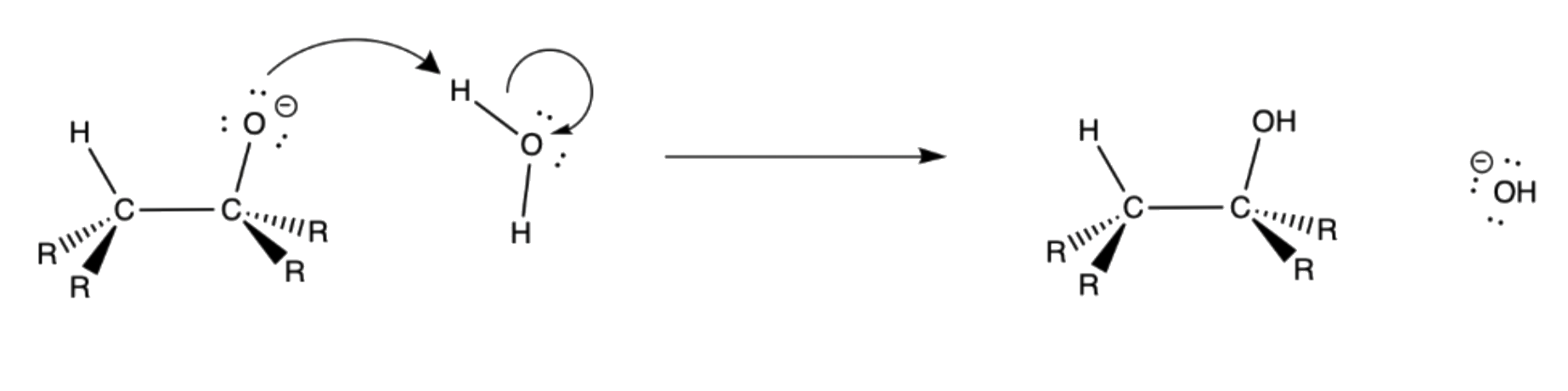
5. A hydroxide ion attacks the empty p-orbital of boron
6. The alkoxide act as the leaving group
The pKaH of hydroxide and alkoxide are similar in value, so they are just as likely to act as the leaving group
We can push the equilibrium to the product side by either adding an excess amount of hydroxide ions or using a polydentate nucleophiles ( with multiple hydroxyl groups ) instead of hydroxide ion for hydrolysis to favor the formation of product due to entropic reasons
Step 5 and 6 are repeated two more times to give three alcohols and a borate ion
Proton transfered from a water moelcule to the alkoxide
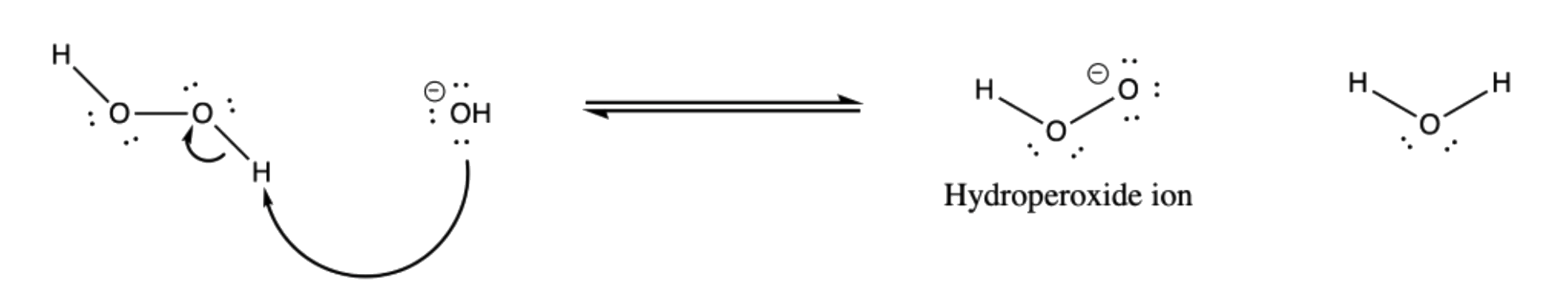
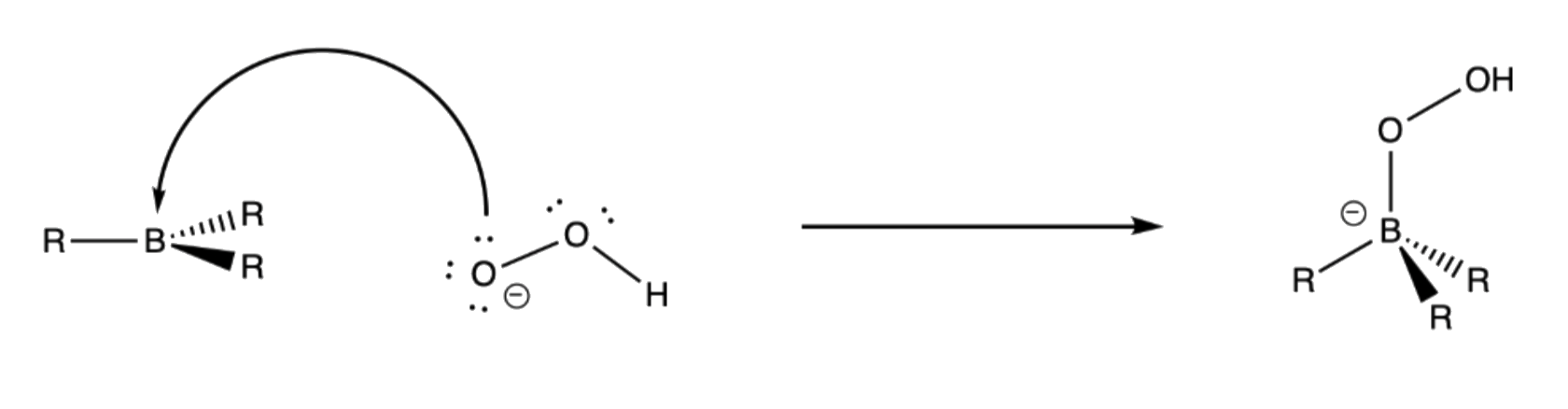
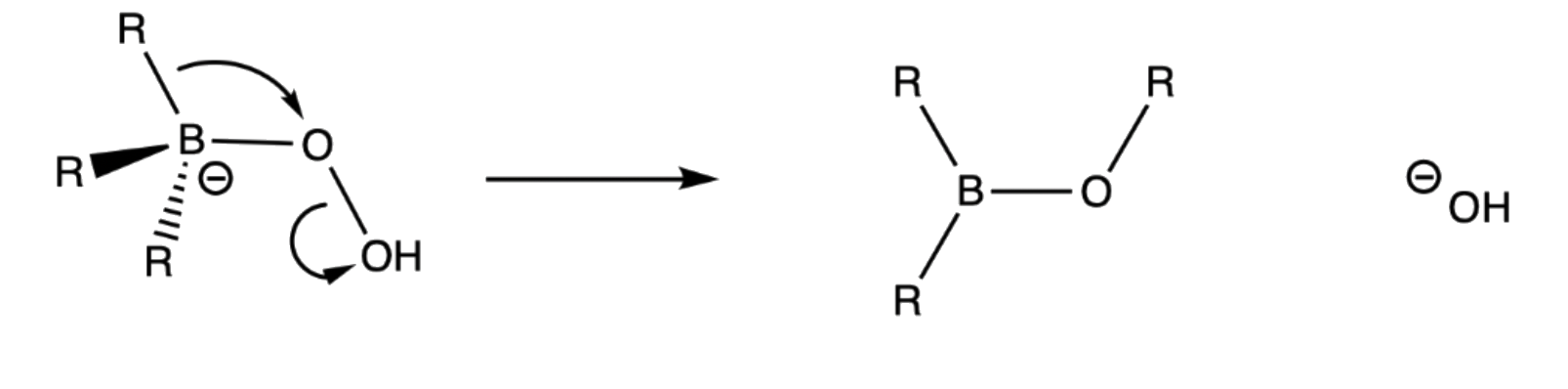
¶ Stereochemistry and Regiochemistry
The addition of boron and hydrogen to the alkene is regioselective and the regioselectivity stems from two reasons
1. Steric Crowding
- The attachment of boron is favored at the less sterically crowded carbon atom of the alkene rather than at the more crowded carbon
2. Stabilization of the partial positive charge on the carbon
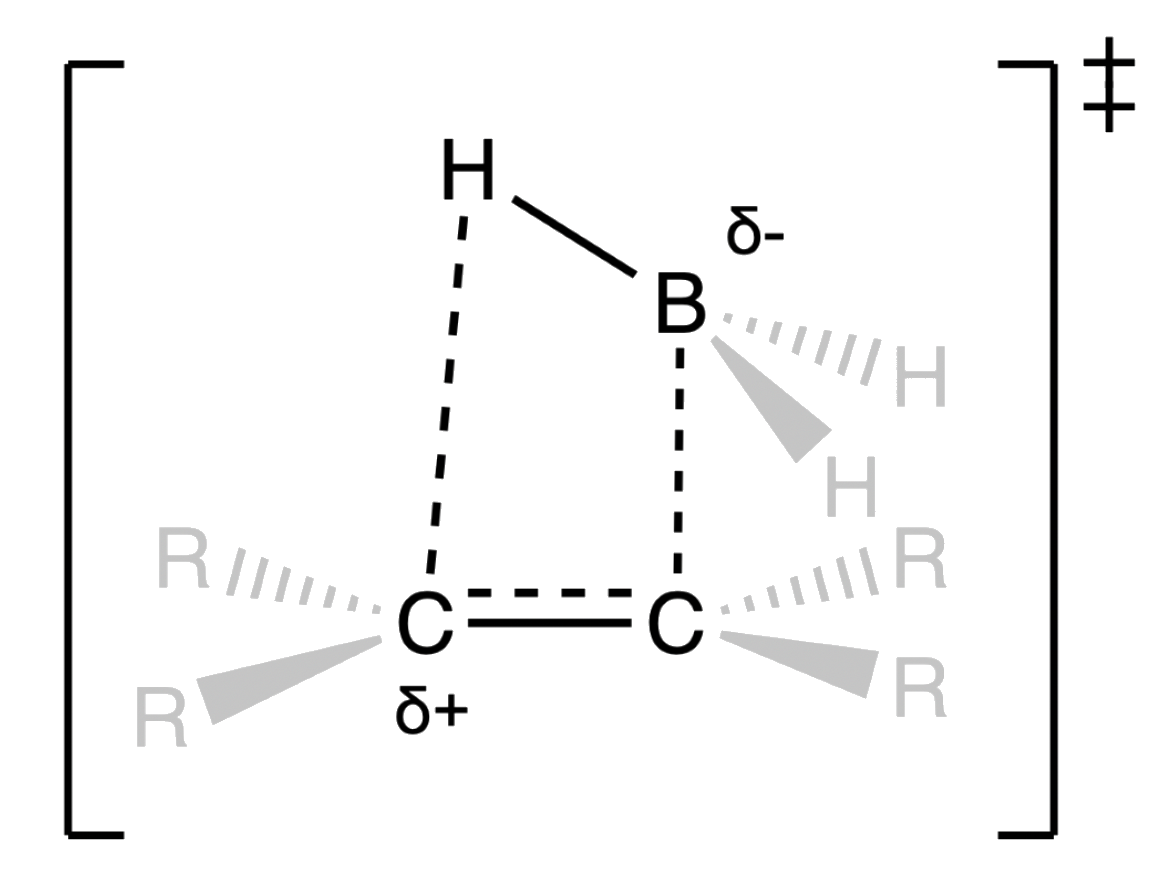
- The addition of the boron and hydrogen to the alkene involves a cyclic, four-centered transition state
- Despite being a concerted step, boron and hydrogen are not added simultaneously
- The C–B bond is formed first, leading to the accumulation of positive charge on the more substituted carbon
- The partial positive charge is like carbocation, so it can be stabilized by a higher degree of substitution
The addition of boron and hydrogen is stereospecific
- The boron and hydrogen are added on the same side of the alkene ( Syn addition )
- Both C–H and C–B bonds form in the same step ( not at the same time )

¶ Syn-Dihydroxylation
Syn-Dihydroxylation adds two hydroxyl group to the same face of an alkene
- The reaction will yield a glycol, aka vicinal diol
- The reaction is an oxidation reaction, so oxidizing agents like osmium tetroxide or permanganate will be used
- In the reaction the oxidizing agents will be consumed
Osmium tetroxide is highly toxic and extremely expensive
- A catalytic amount of the tetroxide is used in the presence of a stochiometric amount of a safe and inexpensive co-oxidant such as N-methylmorpholine N-oxide, abbreviated NMO
- The co-oxidant will oxidize the Os(VI) back to Os(VIII) such that the osmium tetroxide will not be exhausted as long as the co-oxidant is still present
Permanganate is strong a oxidizing agent, so it will continue to oxidize the glycol even after the dihydroxylation reaction is complete
- The reaction is taking place in a cold environment to prevent further oxidation
- The permanganate solution is basified
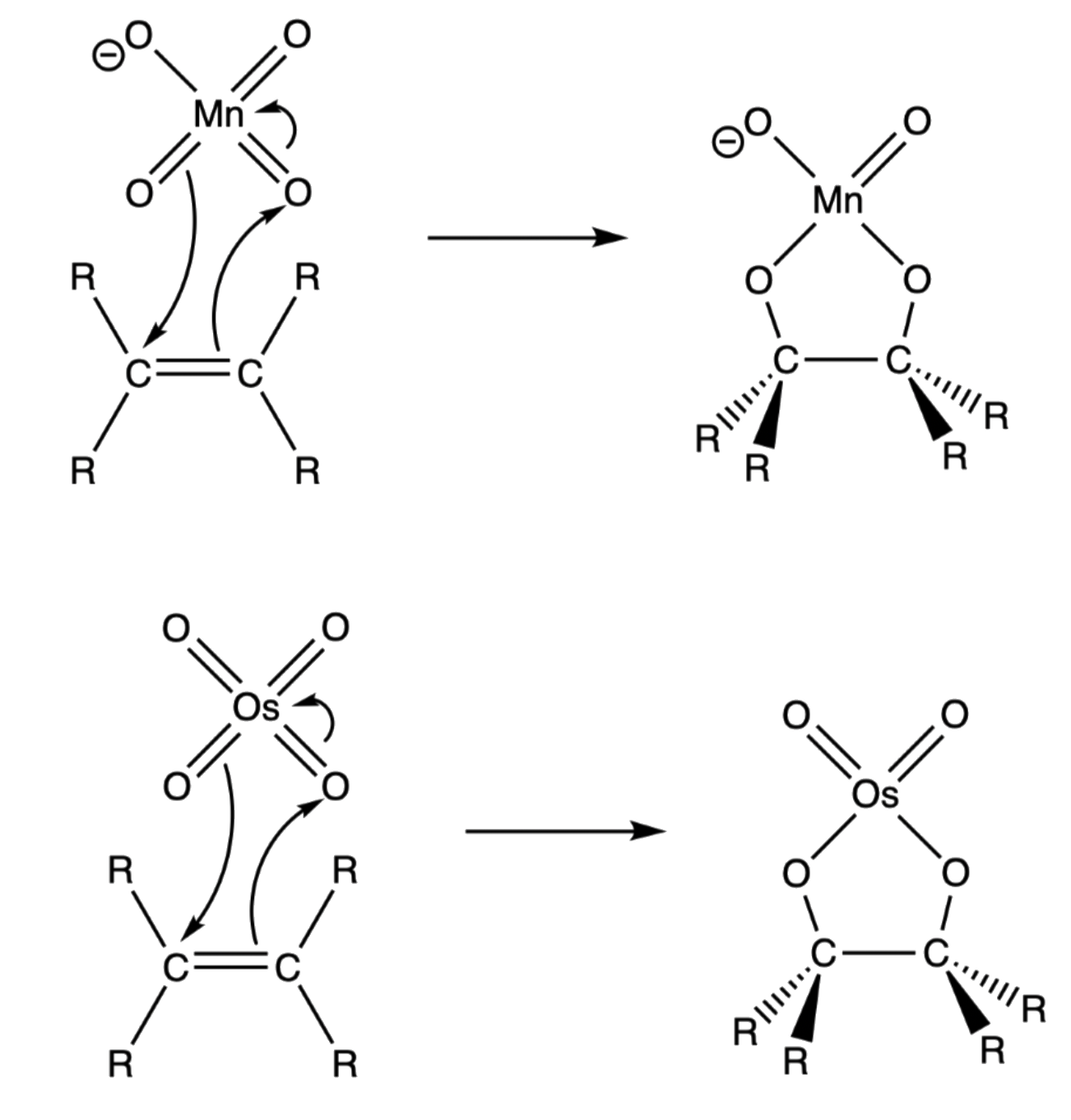
¶ Mechanism
The mechanism is the same for permanganate and osmium tetroxide as they both share a tetrahedral geometry
- A concerted six electron movement to form a cyclic osmate or a cyclic manganate ester
- If permanganate is the chosen reagent, the reacting solution must be cold and basic or else oxidative cleavage will occur
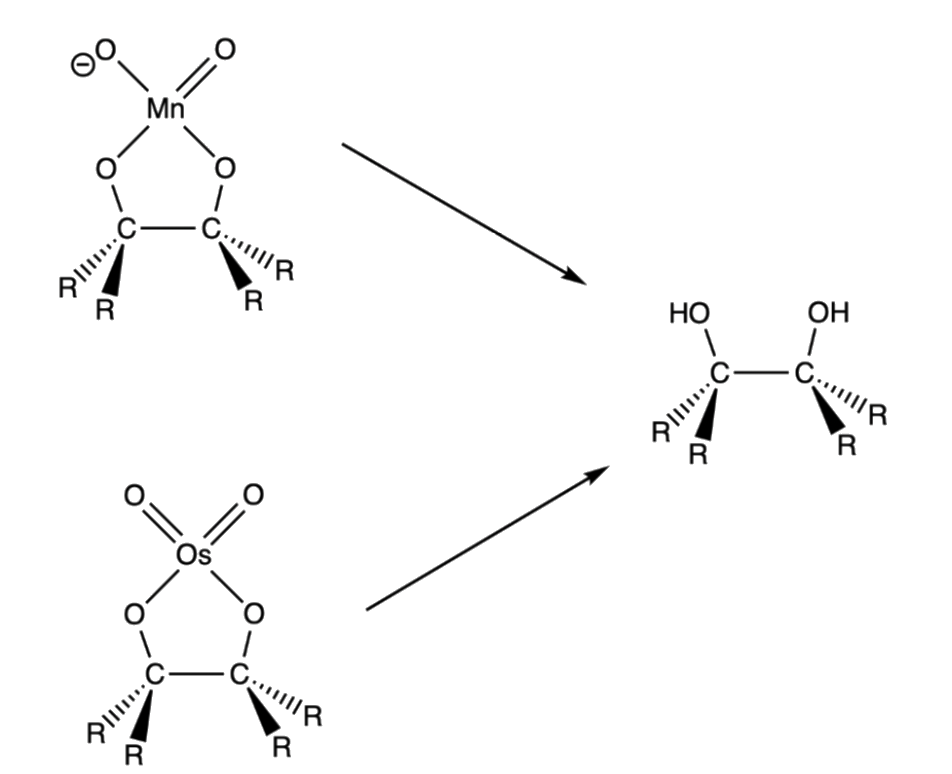
- The intermediate undergo hydrolysis to give the glycol
¶ Stereochemistry and Regiochemistry
The reaction is stereospecific
- It is a syn addition
- The cyclic ester intermediate is formed in such a way that the five-membered osmium/ manganese-containing ring is bonded in a cis configuration to the original alkene
¶ Diel-Alder Reaction
The Diels–Alder reaction is a chemical reaction between a conjugated diene and a dienophile, to form a substituted cyclohexene derivative
- A dienophile is an olefinic or acetylenic component that is seeking a diene in the Diels-Alder reaction
- It is a pericyclic reaction with a concerted mechanism
There are two types of Diel-Alder Reactions: Normal Demand and Inverse Demand
- For the common normal demand reaction, the dienenophile acts as the electrophile while the diene acts as the nucelophile
- For the inverse demand reaction, the dienophile acts as the nucelophile, while the diene acts as the electrophile
In terms of the movement of electrons, both the normal demand and inverse demand reaction has the same mechanism
¶ Mechansim
- The diene adopts the s-cis conformation
-The reaction can only proceed when the diene adopts an s-cis conformation ( left )
- The s-trans conformation is usually favored for steric reasons, so dienes that are locked in the s-cis conformation have a high rate of reaction
- [ 4+ 2 ] oncerted mechanism
- Two carbon–carbon single bonds are formed, resulting in the formation of an unstaturated six-membered ring
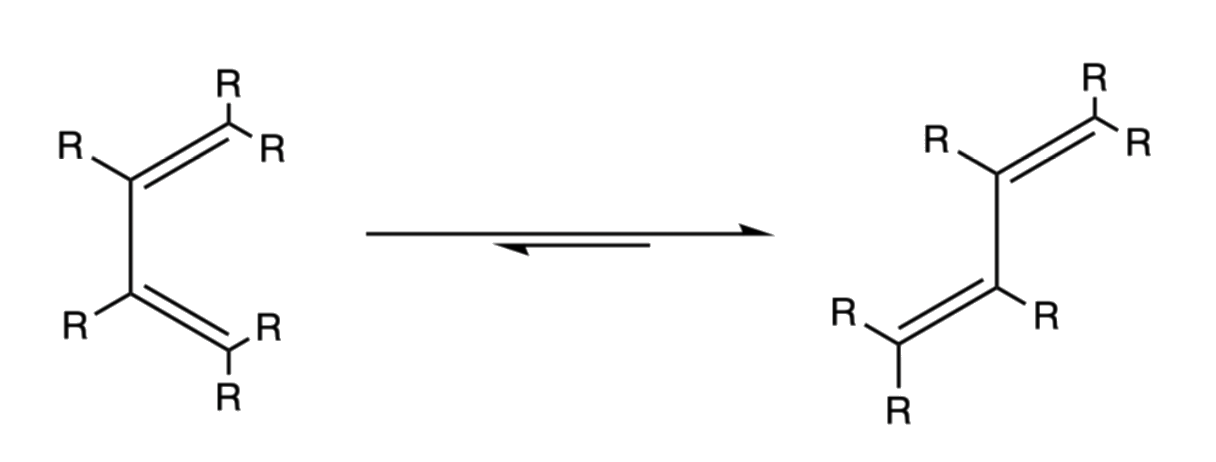
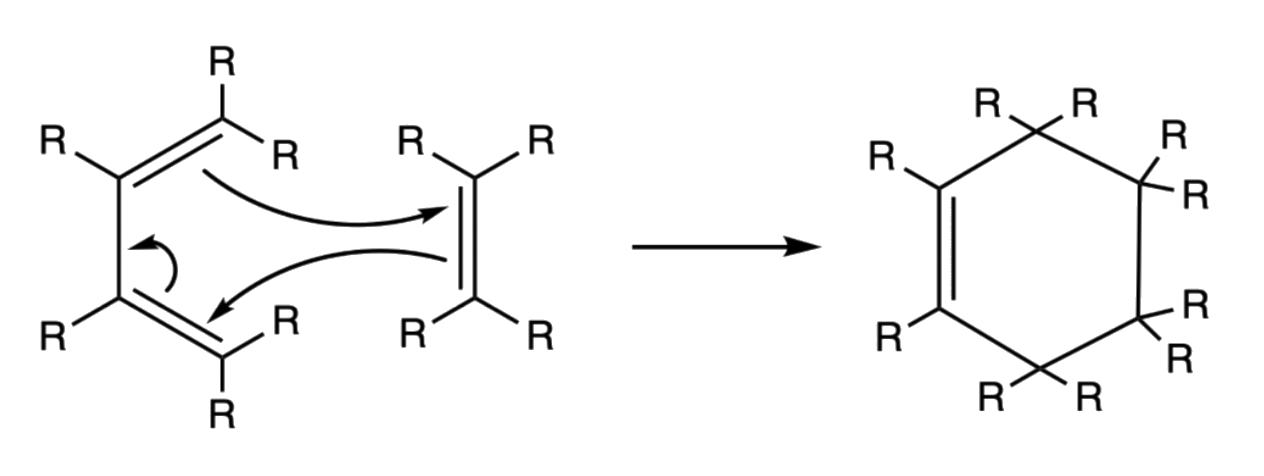
¶ Stereochemistry and Regiochemistry
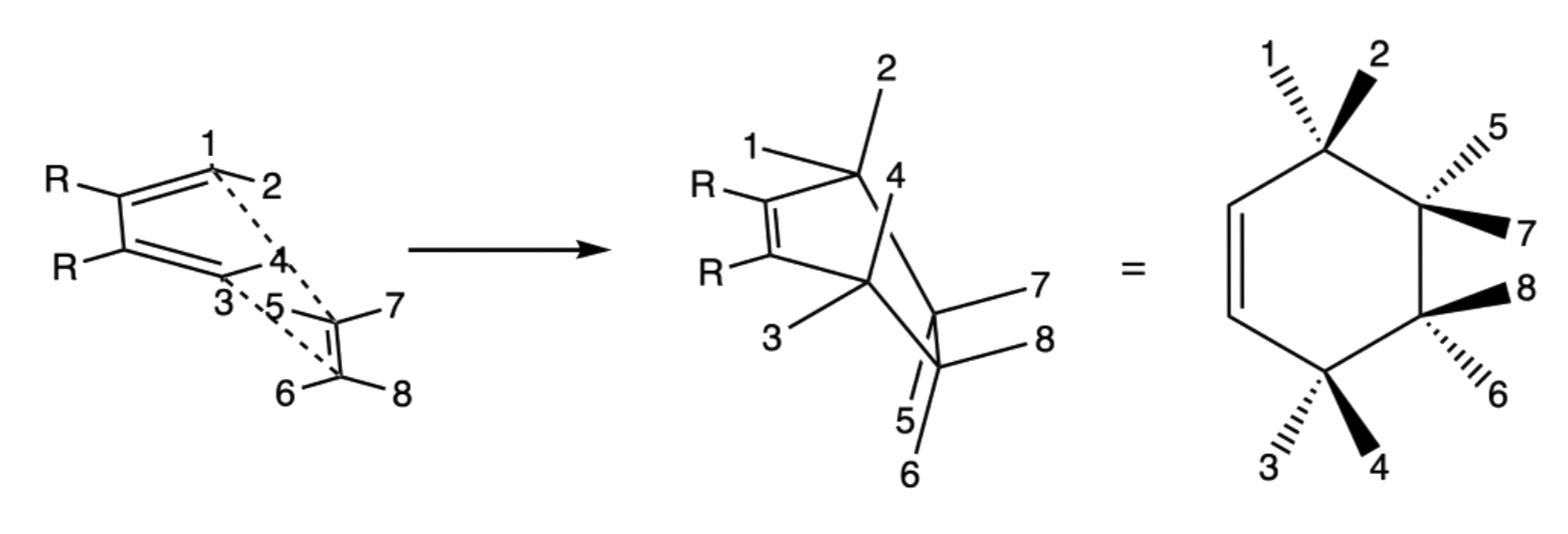
The reaction is stereospecific
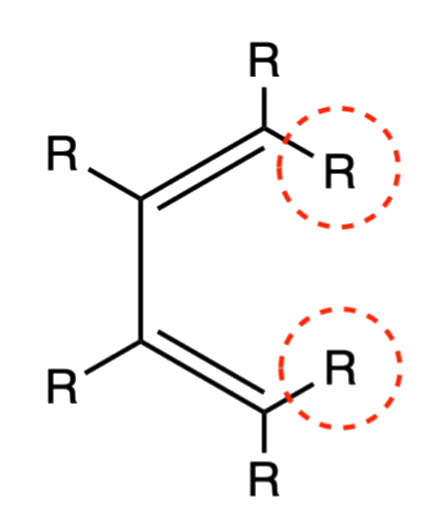
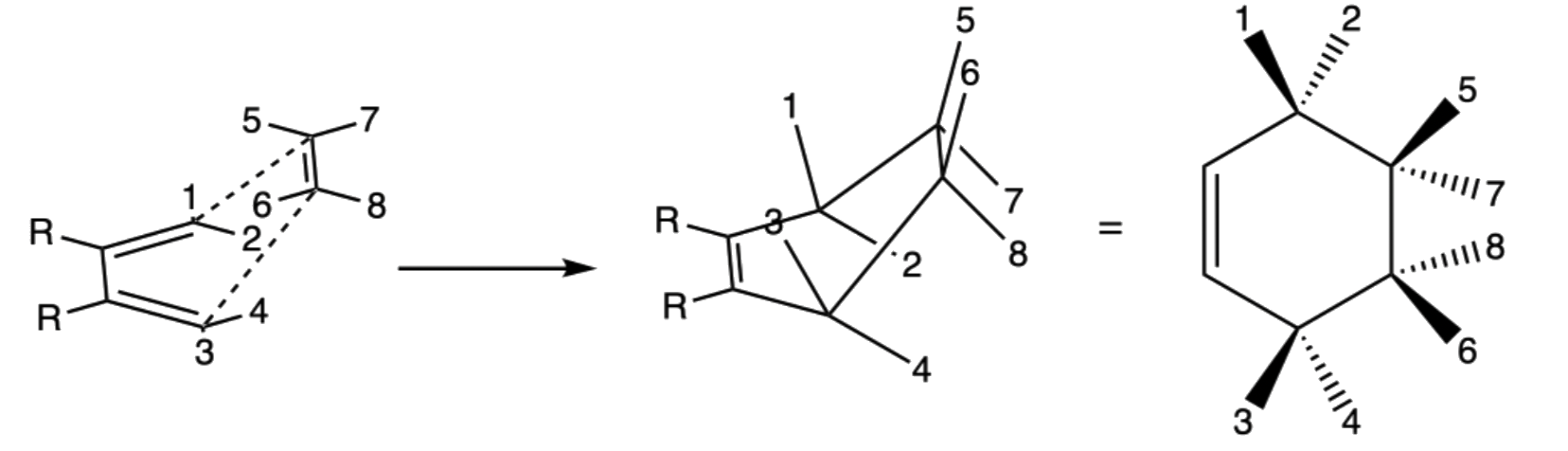
- The relative position of the terminal R groups are retained during the addition as the inner R groups are pushed away to the opposite direction of the approach of the dieneophile
- When the dieneophile attacks from above
- When the dieneophile attacks from below
¶ Catalytic Hydrogenation
Alkene and alkyne react with H2 in the presence of a metal catalyst to convert the π bond(s) into C–H bonds
Catalytic hydrogenation is a heterogenous process
- The reaction does not occur in a homogenous solution but instead on the surface of solid catalyst particles
For hydrogenation of alkyne to alkene, the catalyst must be poisoned such that the alkene formed does not furtehr react to form alkane
¶ Mechanism
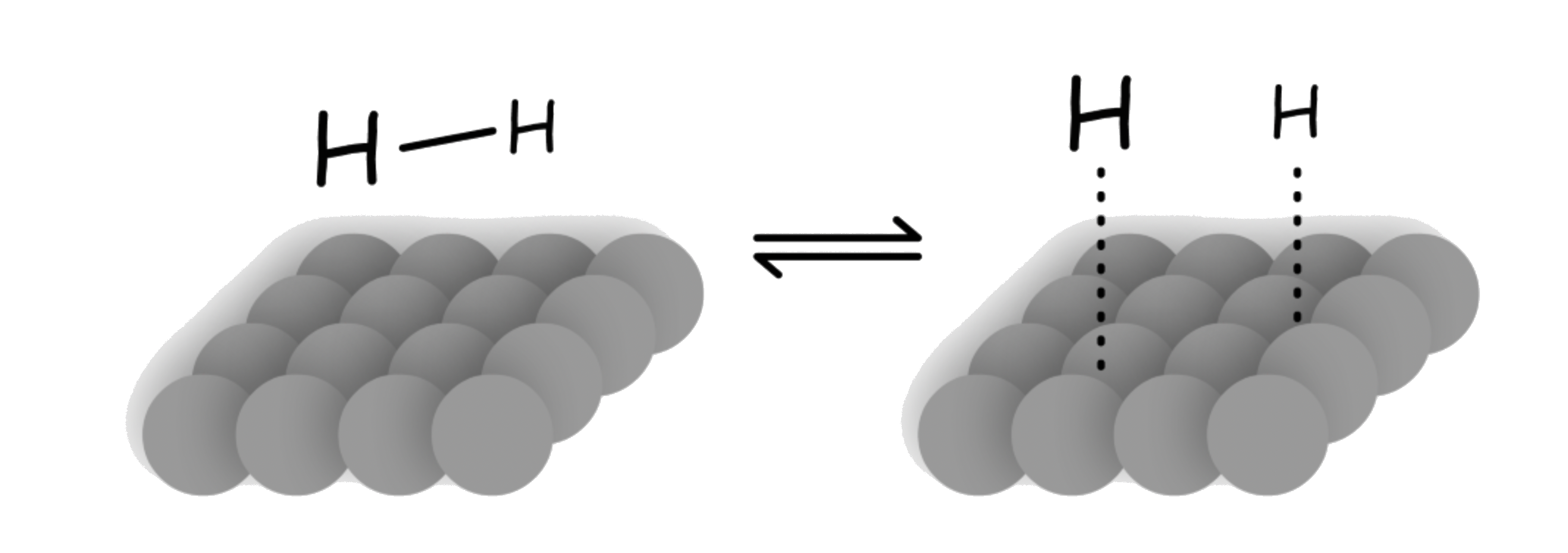
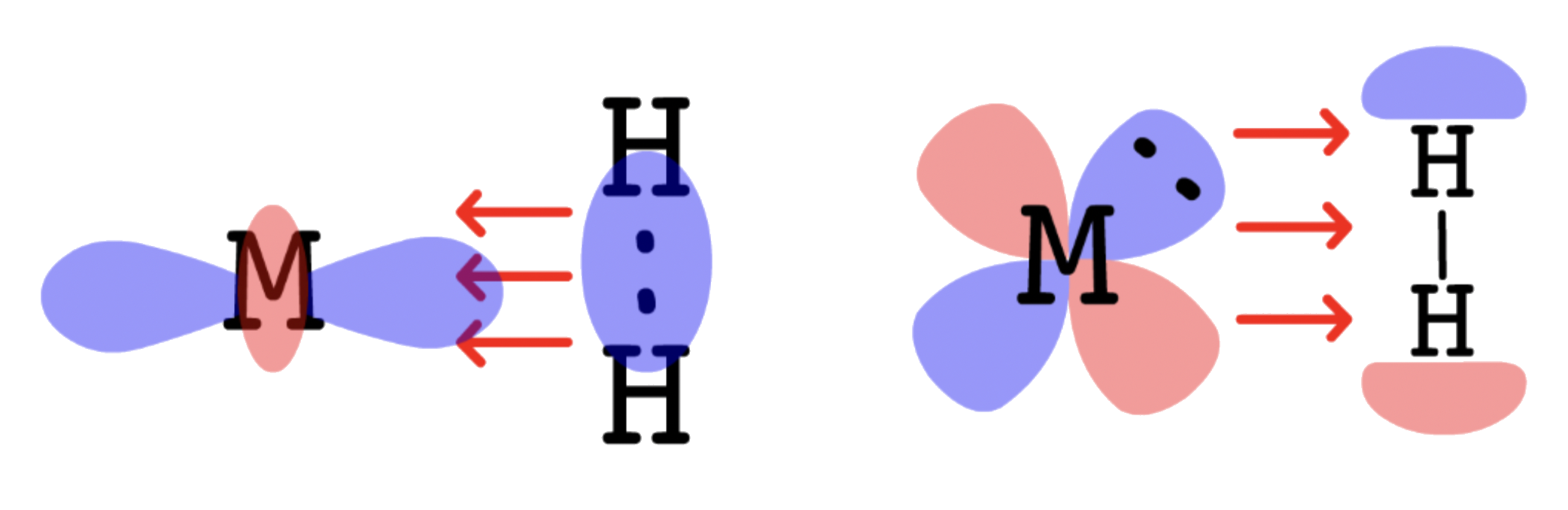
- Molecular hydrogen is adsorbed to the catalyst surface and dissociate into hydrogen atoms
- The H–H bond is broken because electron density of the bonding σ orbital is donated to the metal. At the same time, electron densiry is donated to the σ* orbital of dihydrogen
- The alkene or alkyne is adsorbed to the catalyst surface
- The alkene or alkyne uses their π bond to complex to the metal atoms
- A hydrogen atom is transferred from the metal to the carbon, forming a partially reduced intermediate
- A second hydrogen is transferred from the metal to the second carbon
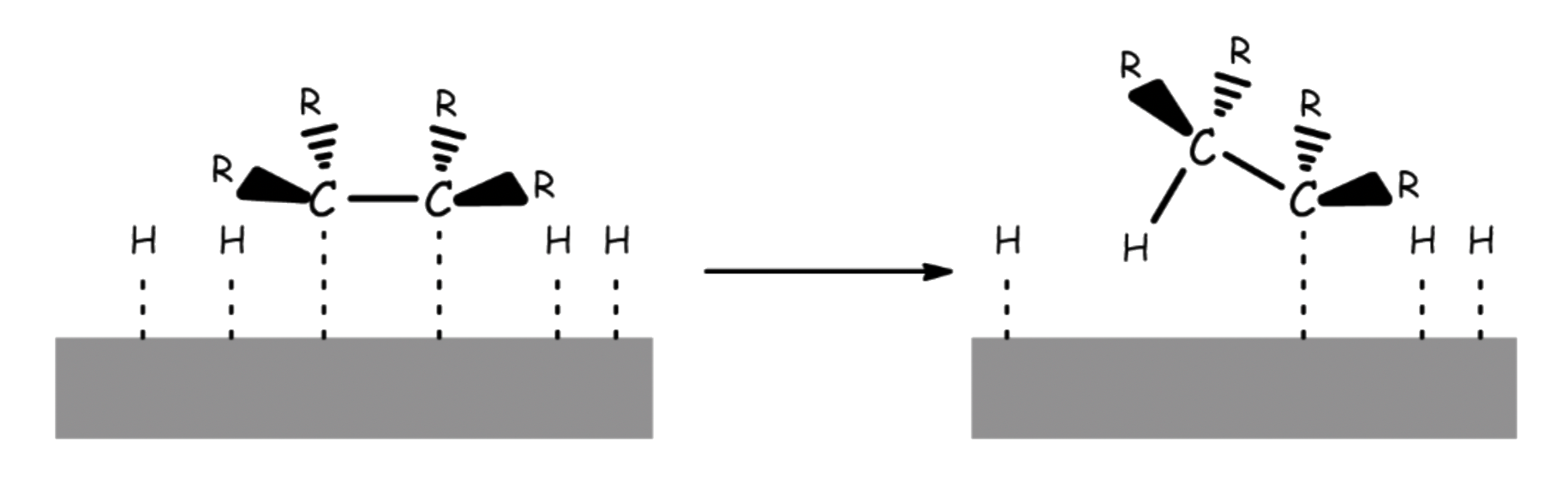
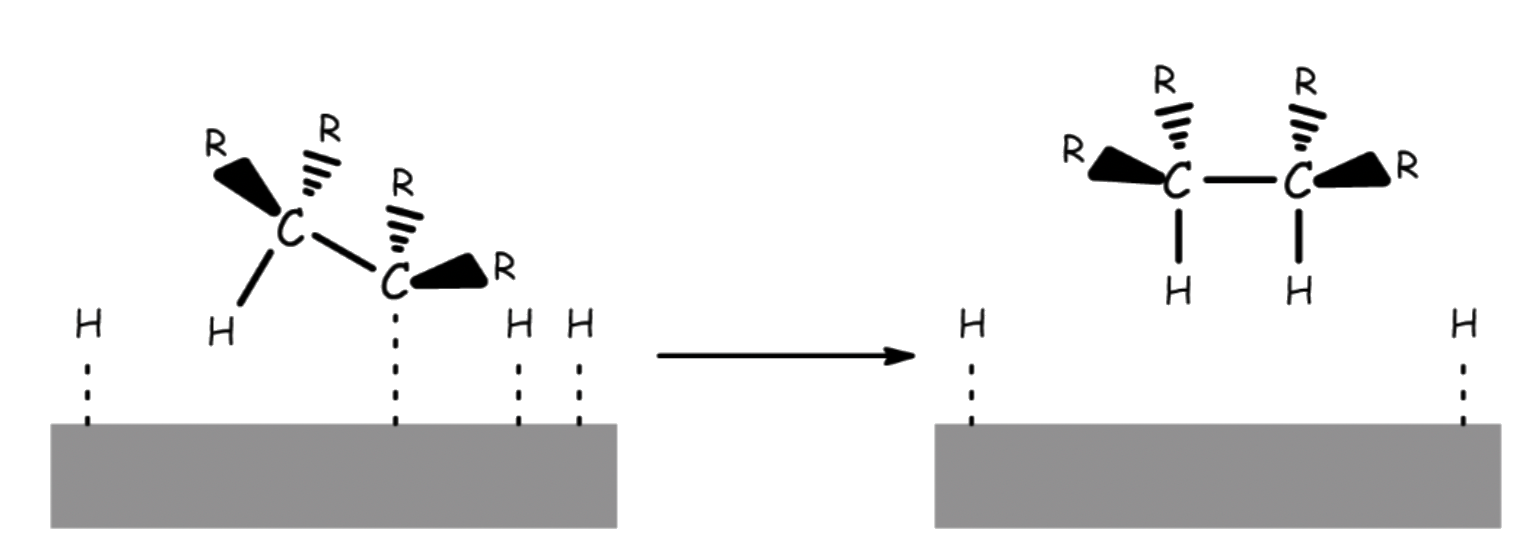
¶ Stereochemistry and Regiochemistry
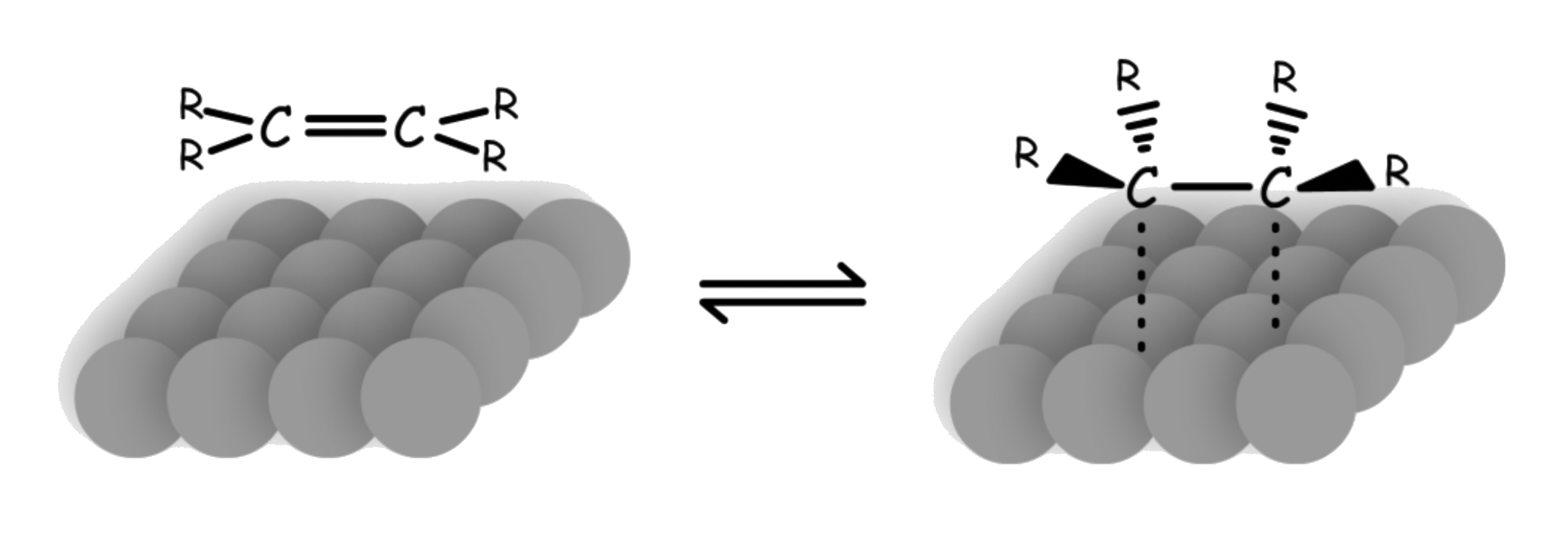
The reaction is stereospecific ( but will produce stereoselective products )
The hydrogen atoms can only be added to the carbon chain across the surface of the metal catalyst
- The two hydrogens are added onto the same face, so it is a syn-addition
- For alkyne, this will lead to the cis-isomer
Under some experimental conditions, particularly with tetrasubstituted double bonds, some percentage of the product appears to be formed by anti-addition of hydrogen
- After both hydrogen is added to the double bond, a hydrogen on a carbon adjacent to the original double bond along with a newly added hydrogen is transferred to the metal surface
- This hydrogen transfer, in effect, reverses the reaction and forms a new alkene that is isomeric with the original alkene
- The newly ( and briefly ) formed alkene is readsorbed and undergoes hydrogenation again. The reaction is still a syn-addition, but the hydrogens are not necessarily added from the same side as the original hydrogen
¶ Birch Reduction
The Birch Reduction, also know as the Dissolving-Metal Reduction, is a hydrogenation reaction
- This hydrogenation reaction can only reduce alkynes, but not alkenes
Alkyne can be reduced to alkenes by using either sodium or lithium metal in liquid ammonia or in low-molecular weight primary or secondary amines
- The alkali metal serves as the reducing agent, while the liquid ammonia or amine acts as the solvent
Alkali metals dissolve in liquid ammonia at –33°C to produce a deep blue solution containing the metal cation and ammonia-solvated electrons
- The ammonia-solvated electrons are free to move around the solution and can react with the alkyne on its own
The Birch Reduction also works on aromatic compounds
¶ Mechanism
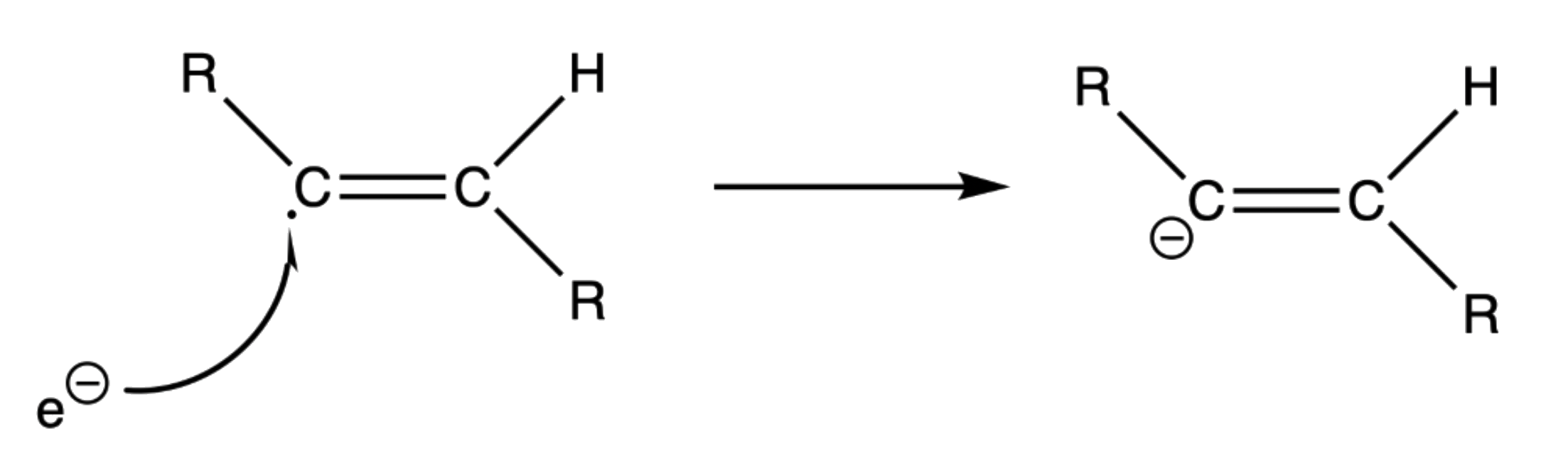
- An electron attacks the triple bond to give an anion radical
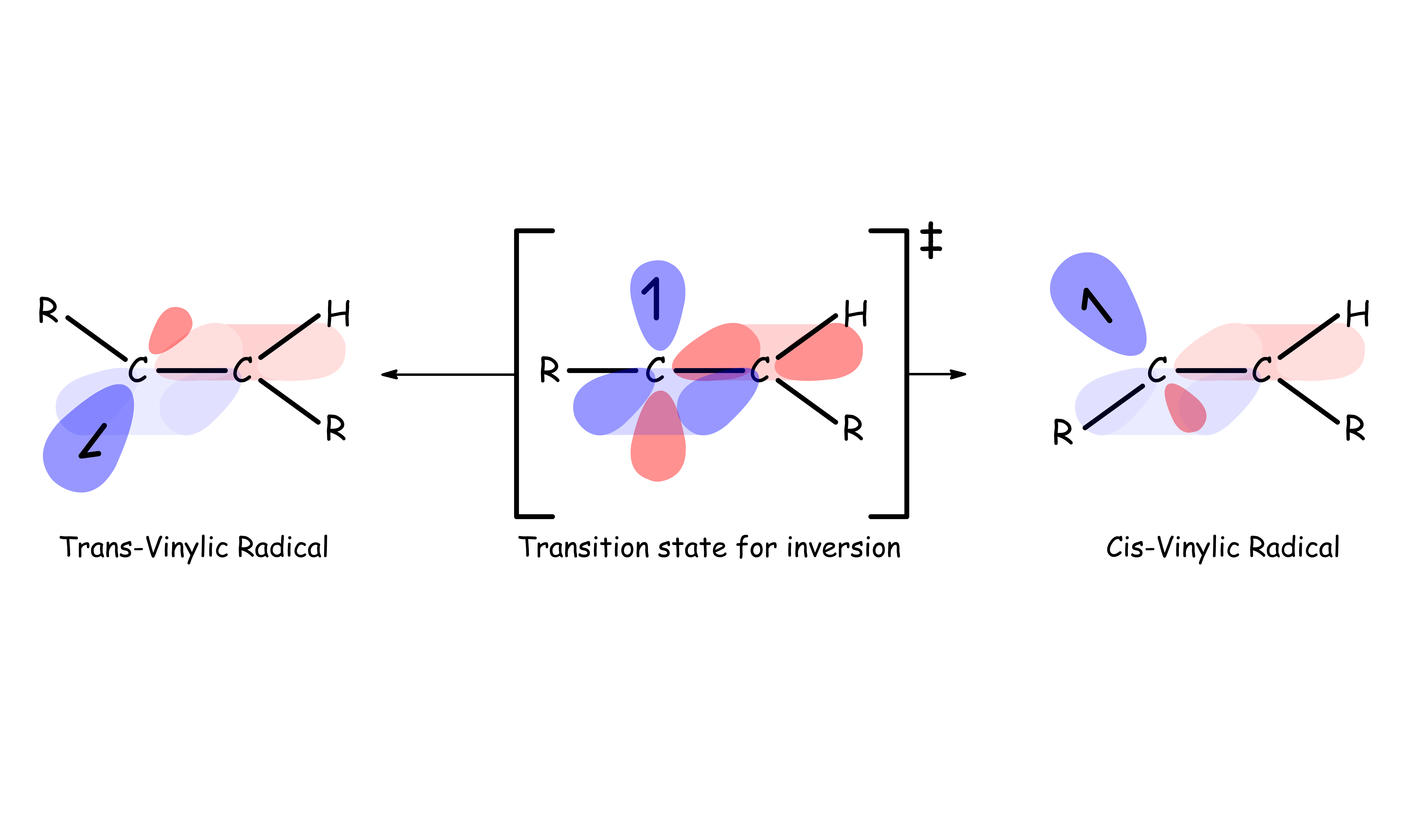
- The radical anion will adopt a trans geometry to reduce steric hinderance
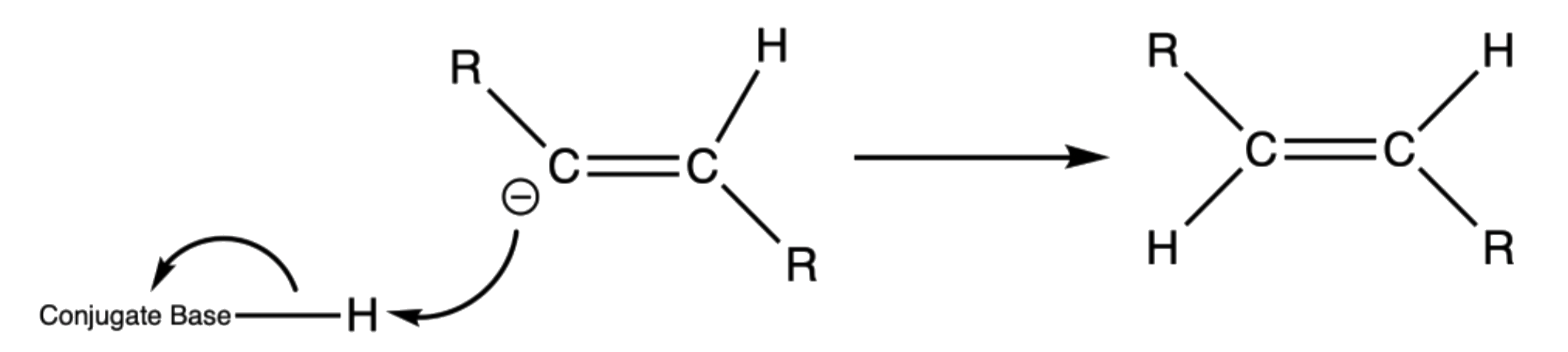
- The anion radical is a strong base, so it will abstract a hydrogen from a nearby acid, usually the solvent, to form a vinylic radical
- The vinyl radical accepts another electron from a second lithium atom to produce a vinyl anion
- The vinyl anion abstracts another proton to form the final trans product
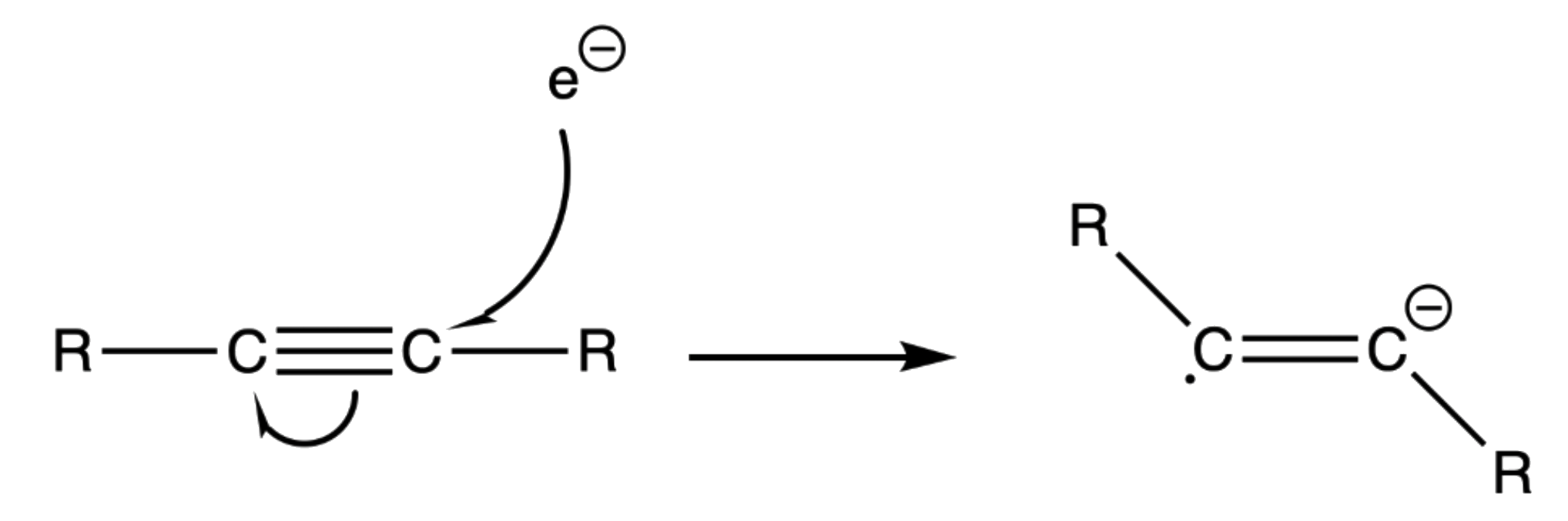
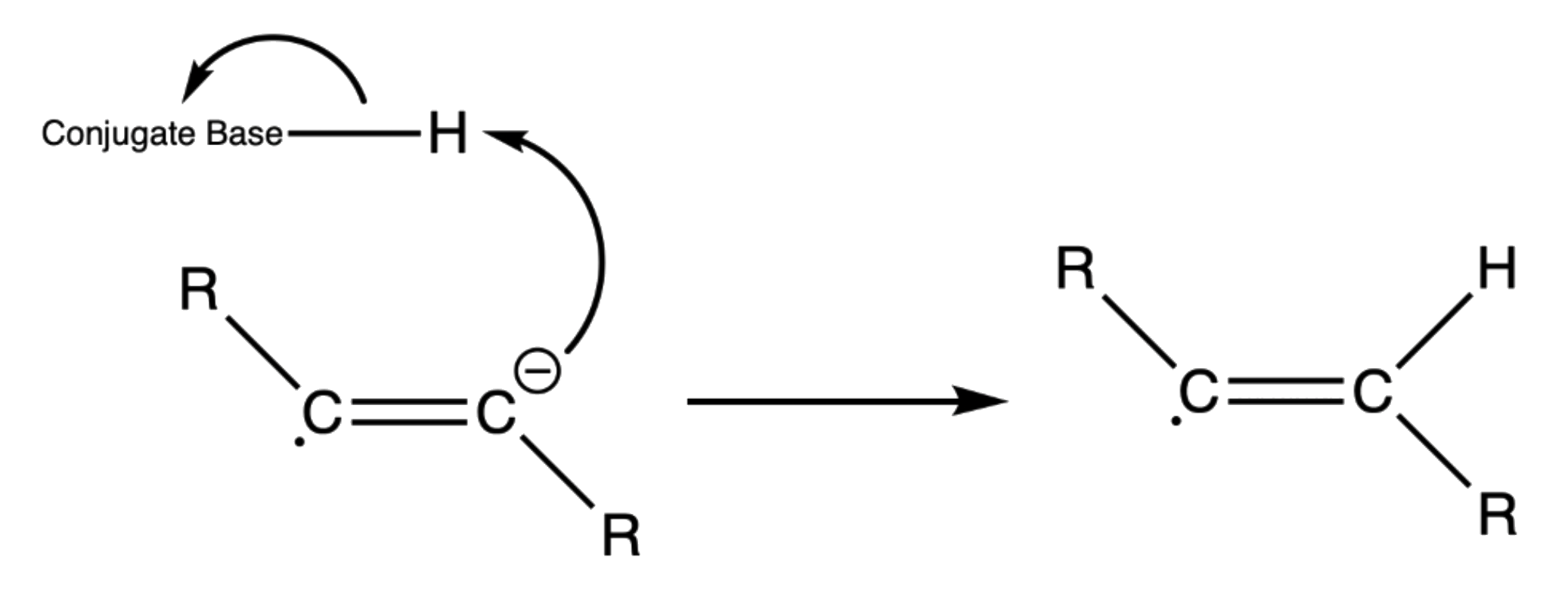
¶ Stereochemistry and Regiochemistry
The reaction is stereoselective
- The hydrogen atoms are added in a trans-fashion since it is more stable for the vinyl radical is more stable when adopting the trans conformation
- It is possible for the vinyl radical to interconvert between the cis and trans conformation through inversion ( not through rotation )
- It is not rotation but inversion from a linear sp form to trigonal planar sp2 that
occurs- The equilibrium position lies toward the trans-vinyl radical due to steric effects